化学信息学驱动的 BACE-1 抑制剂预测:亲和力与分子机理探索
IF 4.3
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
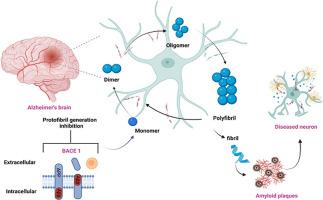
β-分泌酶,有时也称为 BACE1 或 Asp2,是通过分解淀粉样前体蛋白开始生成 Aβ 的酶。因此,BACE 是旨在减少阿尔茨海默病(AD)中 Aβ 生成的药物干预的关键靶点。我们对实验报告为 BACE-1 抑制剂(Ki)的 1235 种化合物进行了定量结构-活性关系(QSAR)研究,以找到与阻断 BACE-1 受体相关的目标分子和结构模式。经合组织(OECD)推荐的遗传算法-多重线性回归(GA-MLR)QSAR 模型在做出准确预测和了解事物的工作原理之间取得了良好的平衡。它在许多评价指标上都达到了较高的数值,包括 R2tr = 0.8047、Q2LMO = 0.802、R2ex = 0.805、CCCex = 0.891、Q2-F1 = 0.801、Q2-F2 = 0.799 和 Q2-F3 = 0.815。通过对 QSAR 的机理解释,我们发现一些氮原子是阻断β-分泌酶并使其无法有效发挥作用的必要条件。带正电荷的芳香族碳原子对 beta-secretase-1 的抑制作用影响更大。对接研究显示,当化合物 27 存在时,催化染料残基 Asp32 和 Asp228 会质子化,但当化合物 27 不存在时,它们会保持非质子化状态。这些吡嗪环和吡啶环与 Tyr71 和 Ile118 残基发生了亲水作用,证实了 QSAR 数据中的药效学特征。我们研究了蛋白质配体复合物和载脂蛋白的稳定性。载脂蛋白的平均回旋半径为 22.13,而蛋白配体复合物的平均回旋半径为 22.91。分子对接、分子动力学(MD)模拟、MMGBSA(分子力学广义博恩表面积)或 DFT 分析的结果均未发生变化。因此,目前的研究工作可有效促进未来药物设计中 BACE 抑制剂的开发。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Cheminformatics-driven prediction of BACE-1 inhibitors: Affinity and molecular mechanism exploration
The β-secretase, sometimes referred to as BACE1 or Asp2, is the enzyme responsible for initiating the production of Aβ by breaking down the amyloid precursor protein. Hence, BACE is a pivotal target for pharmacological intervention aimed at diminishing the production of Aβ in Alzheimer's disease (AD). We did a quantitative structure-activity relationship (QSAR) study on 1235 compounds that had been experimentally reported as BACE-1 inhibitors (Ki) to find the target molecule and structural patterns linked to blocking the BACE-1 receptor. The OECD-recommended genetic algorithm-multiple linear regression (GA-MLR) QSAR model strikes a good balance between being able to make accurate predictions and understanding how things work. It achieves high values for many evaluation metrics, including R2tr = 0.8047, Q2LMO = 0.802, R2ex = 0.805, CCCex = 0.891, Q2-F1=0.801, Q2-F2=0.799, and Q2-F3=0.815. The mechanistic interpretation of QSAR has identified some nitrogen atoms that are required to block beta-secretase and make it less effective at doing its job. A positively charged aromatic carbon atom has a more significant impact on beta-secretase-1 inhibition. The docking study showed that the catalytic dye residues Asp32 and Asp228 become protonated when compound 27 is present, but they stay unprotonated when compound 27 is not present. These rings of pyrazine and pyridine interact hydrophobically with Tyr71 and Ile118 residues, confirming the pharmacophoric features seen in the QSAR data. We examined the stability of both the protein-ligand complex and the apo-protein. The apoprotein had an average gyration radius of 22.13, whereas the protein-ligand complex had an average gyration radius of 22.91. No changes were made to the results from the molecular docking, molecular dynamics (MD) simulation, MMGBSA (Molecular Mechanics Generalized Born Surface Area), or DFT analyses. Therefore, the current work could effectively contribute to the future development of BACE inhibitors in medication design.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Chemical Physics Impact
Materials Science-Materials Science (miscellaneous)
CiteScore
2.60
自引率
0.00%
发文量
65
审稿时长
46 days
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


