{"title":"具有新酚类分子的喹唑啉衍生物的合成:作为替代性多酚氧化酶抑制剂的体外和硅学评估","authors":"Cansu Öztürk, Feyzi Sinan Tokali, Aykut Öztekin, Erbay Kalay, Yalçın Karagöz, Mine Aksoy","doi":"10.1007/s11696-024-03670-5","DOIUrl":null,"url":null,"abstract":"<div><p>Several novel quinazoline derivatives bearing phenolic hydroxyl moiety (2–7) have been produced with good yields and screened for biological activities. All the title compounds were characterized using spectroscopic techniques such as <sup>1</sup>H NMR, <sup>13</sup>C NMR, FTIR, and HRMS. Then, the anti-browning effects of synthesized quinazoline derivatives were investigated in vitro. The IC<sub>50</sub> values for molecules 2–7 were calculated as 0.085, 1.145, 0.106, 6.86, 0.52, 7.07 µM, respectively. K<sub>i</sub> constants, which are inhibitory-enzyme binding constants, were calculated by using Lineweaver–Burk graphs as 0.16 ± 0.0620, 0.906 ± 0.3029, 0.055 ± 0.0171, 9.363 ± 2.5809, 0.773 ± 0.3204, 7.863 ± 1.9107 µM, respectively. In computer-aided analysis, to gain insights electrochemical properties, synthesized compounds were analysed theoretically by density functional theory. Molecular docking studies and MD simulations were performed to identify possible inhibitor-enzyme binding interactions. According to obtained results, all the compounds formed hydrogen bonds with Asn 112 and Asn 414, and showed π-cation interaction with Phe2 70, gatekeeper residue in target protein<i>.</i> Supporting the nm level inhibition, MD simulations indicate that protein-inhibitor complex maintain the stability and have high number of hydrogen bond formation during the simulation.</p></div>","PeriodicalId":513,"journal":{"name":"Chemical Papers","volume":"78 15","pages":"8321 - 8332"},"PeriodicalIF":2.2000,"publicationDate":"2024-09-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Synthesis of quinazoline derivatives with new phenolic moieties: in vitro and in silico evaluations as alternative polyphenol oxidase inhibitors\",\"authors\":\"Cansu Öztürk, Feyzi Sinan Tokali, Aykut Öztekin, Erbay Kalay, Yalçın Karagöz, Mine Aksoy\",\"doi\":\"10.1007/s11696-024-03670-5\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>Several novel quinazoline derivatives bearing phenolic hydroxyl moiety (2–7) have been produced with good yields and screened for biological activities. All the title compounds were characterized using spectroscopic techniques such as <sup>1</sup>H NMR, <sup>13</sup>C NMR, FTIR, and HRMS. Then, the anti-browning effects of synthesized quinazoline derivatives were investigated in vitro. The IC<sub>50</sub> values for molecules 2–7 were calculated as 0.085, 1.145, 0.106, 6.86, 0.52, 7.07 µM, respectively. K<sub>i</sub> constants, which are inhibitory-enzyme binding constants, were calculated by using Lineweaver–Burk graphs as 0.16 ± 0.0620, 0.906 ± 0.3029, 0.055 ± 0.0171, 9.363 ± 2.5809, 0.773 ± 0.3204, 7.863 ± 1.9107 µM, respectively. In computer-aided analysis, to gain insights electrochemical properties, synthesized compounds were analysed theoretically by density functional theory. Molecular docking studies and MD simulations were performed to identify possible inhibitor-enzyme binding interactions. According to obtained results, all the compounds formed hydrogen bonds with Asn 112 and Asn 414, and showed π-cation interaction with Phe2 70, gatekeeper residue in target protein<i>.</i> Supporting the nm level inhibition, MD simulations indicate that protein-inhibitor complex maintain the stability and have high number of hydrogen bond formation during the simulation.</p></div>\",\"PeriodicalId\":513,\"journal\":{\"name\":\"Chemical Papers\",\"volume\":\"78 15\",\"pages\":\"8321 - 8332\"},\"PeriodicalIF\":2.2000,\"publicationDate\":\"2024-09-19\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Chemical Papers\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://link.springer.com/article/10.1007/s11696-024-03670-5\",\"RegionNum\":4,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"Engineering\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Chemical Papers","FirstCategoryId":"92","ListUrlMain":"https://link.springer.com/article/10.1007/s11696-024-03670-5","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"Engineering","Score":null,"Total":0}
Synthesis of quinazoline derivatives with new phenolic moieties: in vitro and in silico evaluations as alternative polyphenol oxidase inhibitors
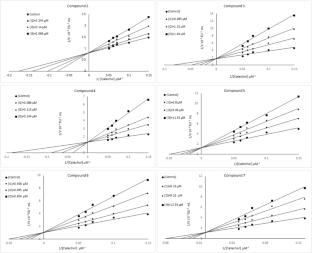
Several novel quinazoline derivatives bearing phenolic hydroxyl moiety (2–7) have been produced with good yields and screened for biological activities. All the title compounds were characterized using spectroscopic techniques such as 1H NMR, 13C NMR, FTIR, and HRMS. Then, the anti-browning effects of synthesized quinazoline derivatives were investigated in vitro. The IC50 values for molecules 2–7 were calculated as 0.085, 1.145, 0.106, 6.86, 0.52, 7.07 µM, respectively. Ki constants, which are inhibitory-enzyme binding constants, were calculated by using Lineweaver–Burk graphs as 0.16 ± 0.0620, 0.906 ± 0.3029, 0.055 ± 0.0171, 9.363 ± 2.5809, 0.773 ± 0.3204, 7.863 ± 1.9107 µM, respectively. In computer-aided analysis, to gain insights electrochemical properties, synthesized compounds were analysed theoretically by density functional theory. Molecular docking studies and MD simulations were performed to identify possible inhibitor-enzyme binding interactions. According to obtained results, all the compounds formed hydrogen bonds with Asn 112 and Asn 414, and showed π-cation interaction with Phe2 70, gatekeeper residue in target protein. Supporting the nm level inhibition, MD simulations indicate that protein-inhibitor complex maintain the stability and have high number of hydrogen bond formation during the simulation.
Chemical PapersChemical Engineering-General Chemical Engineering
CiteScore
3.30
自引率
4.50%
发文量
590
期刊介绍:
Chemical Papers is a peer-reviewed, international journal devoted to basic and applied chemical research. It has a broad scope covering the chemical sciences, but favors interdisciplinary research and studies that bring chemistry together with other disciplines.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


