Camille Chartier, Rana Deeba, Alexandra Collard, Sylvie Chardon-Noblat and Cyrille Costentin*,
{"title":"四苯基卟啉铁(I)是一种活性催化剂,可在从 CO2 到等电子 N2O 的还原脱氧反应中发挥作用","authors":"Camille Chartier, Rana Deeba, Alexandra Collard, Sylvie Chardon-Noblat and Cyrille Costentin*, ","doi":"10.1021/acscatal.4c0525910.1021/acscatal.4c05259","DOIUrl":null,"url":null,"abstract":"<p >The electrochemical reductive deoxygenation of N<sub>2</sub>O catalyzed by iron tetraphenylporphyrin (TPPFe) is studied and compared to the electroreduction of CO<sub>2</sub> to CO by the same catalyst. We show that the electroreduction of N<sub>2</sub>O is catalyzed by TPPFe(I), albeit at a slower rate compared to that of TPPFe(0). On a similar time scale, CO<sub>2</sub> does not react with TPPFe(I). The catalytic reduction of N<sub>2</sub>O by TPPFe(I) is however endowed by a self-modulation process due to the production of hydroxide ions as a coproduct that bind to TPPFe(II) and slow down the regeneration of the TPPFe(I) catalytic active species. Two catalytic cycles are thus intertwined when the electrocatalysis is run at a potential where TPPFe(0) is generated, and the resting state in solution is TPPFe(II)(OH) and not TPPFe(I), as opposed to the case of CO<sub>2</sub> catalytic reductive deoxygenation. Revealing the shift of the active catalysis from TPPFe(0) to TPPFe(I) opens the way toward the design of molecular catalysts for N<sub>2</sub>O reductive deoxygenation at a lower overpotential.</p>","PeriodicalId":9,"journal":{"name":"ACS Catalysis ","volume":"14 19","pages":"14509–14516 14509–14516"},"PeriodicalIF":13.1000,"publicationDate":"2024-09-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Iron(I) Tetraphenylporphyrin Is an Active Catalyst in Reductive Deoxygenation when Switching from CO2 to Isoelectronic N2O\",\"authors\":\"Camille Chartier, Rana Deeba, Alexandra Collard, Sylvie Chardon-Noblat and Cyrille Costentin*, \",\"doi\":\"10.1021/acscatal.4c0525910.1021/acscatal.4c05259\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >The electrochemical reductive deoxygenation of N<sub>2</sub>O catalyzed by iron tetraphenylporphyrin (TPPFe) is studied and compared to the electroreduction of CO<sub>2</sub> to CO by the same catalyst. We show that the electroreduction of N<sub>2</sub>O is catalyzed by TPPFe(I), albeit at a slower rate compared to that of TPPFe(0). On a similar time scale, CO<sub>2</sub> does not react with TPPFe(I). The catalytic reduction of N<sub>2</sub>O by TPPFe(I) is however endowed by a self-modulation process due to the production of hydroxide ions as a coproduct that bind to TPPFe(II) and slow down the regeneration of the TPPFe(I) catalytic active species. Two catalytic cycles are thus intertwined when the electrocatalysis is run at a potential where TPPFe(0) is generated, and the resting state in solution is TPPFe(II)(OH) and not TPPFe(I), as opposed to the case of CO<sub>2</sub> catalytic reductive deoxygenation. Revealing the shift of the active catalysis from TPPFe(0) to TPPFe(I) opens the way toward the design of molecular catalysts for N<sub>2</sub>O reductive deoxygenation at a lower overpotential.</p>\",\"PeriodicalId\":9,\"journal\":{\"name\":\"ACS Catalysis \",\"volume\":\"14 19\",\"pages\":\"14509–14516 14509–14516\"},\"PeriodicalIF\":13.1000,\"publicationDate\":\"2024-09-16\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"ACS Catalysis \",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acscatal.4c05259\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"ACS Catalysis ","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acscatal.4c05259","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
摘要
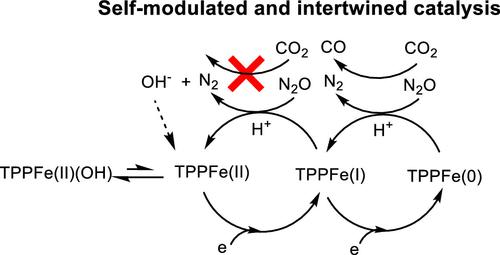
我们研究了四苯基卟啉铁(TPPFe)催化的 N2O 电化学还原脱氧反应,并与同一催化剂催化 CO2 电还原为 CO 的反应进行了比较。我们发现,四苯基卟啉铁(TPPFe(I))可催化 N2O 的电还原,尽管与四苯基卟啉铁(TPPFe(0))相比速度较慢。在类似的时间范围内,二氧化碳不会与 TPPFe(I) 发生反应。不过,TPPFe(I) 催化还原 N2O 的过程具有自我调节功能,因为会产生氢氧根离子作为副产物,与 TPPFe(II) 结合,减缓 TPPFe(I) 催化活性物质的再生速度。因此,当电催化在产生 TPPFe(0) 的电位下运行时,两个催化循环交织在一起,溶液中的静止状态是 TPPFe(II)(OH),而不是 TPPFe(I),这与 CO2 催化还原脱氧的情况不同。揭示活性催化作用从 TPPFe(0) 到 TPPFe(I) 的转变,为设计过电势较低的 N2O 还原脱氧分子催化剂开辟了道路。
Iron(I) Tetraphenylporphyrin Is an Active Catalyst in Reductive Deoxygenation when Switching from CO2 to Isoelectronic N2O
The electrochemical reductive deoxygenation of N2O catalyzed by iron tetraphenylporphyrin (TPPFe) is studied and compared to the electroreduction of CO2 to CO by the same catalyst. We show that the electroreduction of N2O is catalyzed by TPPFe(I), albeit at a slower rate compared to that of TPPFe(0). On a similar time scale, CO2 does not react with TPPFe(I). The catalytic reduction of N2O by TPPFe(I) is however endowed by a self-modulation process due to the production of hydroxide ions as a coproduct that bind to TPPFe(II) and slow down the regeneration of the TPPFe(I) catalytic active species. Two catalytic cycles are thus intertwined when the electrocatalysis is run at a potential where TPPFe(0) is generated, and the resting state in solution is TPPFe(II)(OH) and not TPPFe(I), as opposed to the case of CO2 catalytic reductive deoxygenation. Revealing the shift of the active catalysis from TPPFe(0) to TPPFe(I) opens the way toward the design of molecular catalysts for N2O reductive deoxygenation at a lower overpotential.
期刊介绍:
ACS Catalysis is an esteemed journal that publishes original research in the fields of heterogeneous catalysis, molecular catalysis, and biocatalysis. It offers broad coverage across diverse areas such as life sciences, organometallics and synthesis, photochemistry and electrochemistry, drug discovery and synthesis, materials science, environmental protection, polymer discovery and synthesis, and energy and fuels.
The scope of the journal is to showcase innovative work in various aspects of catalysis. This includes new reactions and novel synthetic approaches utilizing known catalysts, the discovery or modification of new catalysts, elucidation of catalytic mechanisms through cutting-edge investigations, practical enhancements of existing processes, as well as conceptual advances in the field. Contributions to ACS Catalysis can encompass both experimental and theoretical research focused on catalytic molecules, macromolecules, and materials that exhibit catalytic turnover.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


