Chandra Shahi*, Rohan Maniar, Jinliang Ning, Gábor I. Csonka, John P. Perdew* and Adrienn Ruzsinszky*,
{"title":"垂直电离能、广义科恩-沙姆轨道能和氧化铜阴离子的奇特情况","authors":"Chandra Shahi*, Rohan Maniar, Jinliang Ning, Gábor I. Csonka, John P. Perdew* and Adrienn Ruzsinszky*, ","doi":"10.1021/acs.jpca.4c0364010.1021/acs.jpca.4c03640","DOIUrl":null,"url":null,"abstract":"<p >Are the vertical ionization energies from a bound electronic system, initially in its ground state, equal to minus the corresponding exact Kohn–Sham orbital energies of density functional theory (DFT)? This is known to be true for the first or lowest vertical ionization energy. We show that the correction from time-dependent DFT arises from the continuum and need not vanish. Recent work compared the experimental photoemission thresholds of the molecules Cu<sub>2</sub>O<sup>–</sup>, CuO<sup>–</sup>, CuO<sub>2</sub><sup>–</sup>, and CuO<sub>3</sub><sup>–</sup> with minus the corresponding orbital energies from a generalized gradient approximation (GGA) and its global and range-separated hybrids with exact exchange, finding striking differences which were attributed to self-interaction error, strong correlation, or both. Here, we extend that work to include the local spin density approximation (LSDA), its Perdew–Zunger self-interaction correction with Fermi–Löwdin localized orbitals (LSDA-SIC), a quasi-self-consistent locally scaled-down version of LSDA-SIC (QLSIC), and the Quantum Theory Project QTP02 range-separated hybrid functional, all but LSDA implemented in a generalized Kohn–Sham approach. QTP02 impressively yields a near equality for many sp-bonded molecules. However, for the copper oxide anions studied here, none of the tested methods reproduces the experimental photoemission thresholds.</p>","PeriodicalId":59,"journal":{"name":"The Journal of Physical Chemistry A","volume":"128 40","pages":"8628–8634 8628–8634"},"PeriodicalIF":2.8000,"publicationDate":"2024-09-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Vertical Ionization Energies, Generalized Kohn–Sham Orbital Energies, and the Curious Case of the Copper Oxide Anions\",\"authors\":\"Chandra Shahi*, Rohan Maniar, Jinliang Ning, Gábor I. Csonka, John P. Perdew* and Adrienn Ruzsinszky*, \",\"doi\":\"10.1021/acs.jpca.4c0364010.1021/acs.jpca.4c03640\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Are the vertical ionization energies from a bound electronic system, initially in its ground state, equal to minus the corresponding exact Kohn–Sham orbital energies of density functional theory (DFT)? This is known to be true for the first or lowest vertical ionization energy. We show that the correction from time-dependent DFT arises from the continuum and need not vanish. Recent work compared the experimental photoemission thresholds of the molecules Cu<sub>2</sub>O<sup>–</sup>, CuO<sup>–</sup>, CuO<sub>2</sub><sup>–</sup>, and CuO<sub>3</sub><sup>–</sup> with minus the corresponding orbital energies from a generalized gradient approximation (GGA) and its global and range-separated hybrids with exact exchange, finding striking differences which were attributed to self-interaction error, strong correlation, or both. Here, we extend that work to include the local spin density approximation (LSDA), its Perdew–Zunger self-interaction correction with Fermi–Löwdin localized orbitals (LSDA-SIC), a quasi-self-consistent locally scaled-down version of LSDA-SIC (QLSIC), and the Quantum Theory Project QTP02 range-separated hybrid functional, all but LSDA implemented in a generalized Kohn–Sham approach. QTP02 impressively yields a near equality for many sp-bonded molecules. However, for the copper oxide anions studied here, none of the tested methods reproduces the experimental photoemission thresholds.</p>\",\"PeriodicalId\":59,\"journal\":{\"name\":\"The Journal of Physical Chemistry A\",\"volume\":\"128 40\",\"pages\":\"8628–8634 8628–8634\"},\"PeriodicalIF\":2.8000,\"publicationDate\":\"2024-09-27\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"The Journal of Physical Chemistry A\",\"FirstCategoryId\":\"1\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jpca.4c03640\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry A","FirstCategoryId":"1","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jpca.4c03640","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Vertical Ionization Energies, Generalized Kohn–Sham Orbital Energies, and the Curious Case of the Copper Oxide Anions
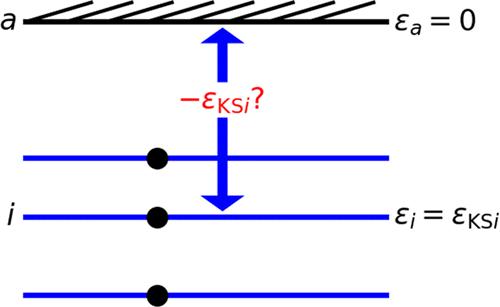
Are the vertical ionization energies from a bound electronic system, initially in its ground state, equal to minus the corresponding exact Kohn–Sham orbital energies of density functional theory (DFT)? This is known to be true for the first or lowest vertical ionization energy. We show that the correction from time-dependent DFT arises from the continuum and need not vanish. Recent work compared the experimental photoemission thresholds of the molecules Cu2O–, CuO–, CuO2–, and CuO3– with minus the corresponding orbital energies from a generalized gradient approximation (GGA) and its global and range-separated hybrids with exact exchange, finding striking differences which were attributed to self-interaction error, strong correlation, or both. Here, we extend that work to include the local spin density approximation (LSDA), its Perdew–Zunger self-interaction correction with Fermi–Löwdin localized orbitals (LSDA-SIC), a quasi-self-consistent locally scaled-down version of LSDA-SIC (QLSIC), and the Quantum Theory Project QTP02 range-separated hybrid functional, all but LSDA implemented in a generalized Kohn–Sham approach. QTP02 impressively yields a near equality for many sp-bonded molecules. However, for the copper oxide anions studied here, none of the tested methods reproduces the experimental photoemission thresholds.
期刊介绍:
The Journal of Physical Chemistry A is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


