Colin M. Hylton-Farrington, and , Richard C. Remsing*,
{"title":"卤化物过氧化物中电子密度的动态局部对称波动","authors":"Colin M. Hylton-Farrington, and , Richard C. Remsing*, ","doi":"10.1021/acs.chemmater.4c0112110.1021/acs.chemmater.4c01121","DOIUrl":null,"url":null,"abstract":"<p >Metal halide perovskites have emerged as an exciting class of materials for applications in solar energy harvesting, optical devices, catalysis, and other photophysical applications. Many of the exciting properties of halide perovskites are tied to their soft, dynamic, and anharmonic lattice. In particular, the precise coupling between anharmonic lattice dynamics and electronic fluctuations is not completely understood. To build an understanding of this coupling, we use ab initio molecular dynamics simulations supplemented by the calculation of maximally localized Wannier functions to carry out a dynamic group theory analysis of local electron density fluctuations and how these fluctuations are coupled to lattice fluctuations in the model inorganic halide perovskite CsSnBr<sub>3</sub>. We detail symmetry-dependent couplings between vibrational modes including octahedral tilting. Importantly, we suggest that the large anharmonicity of some of the vibrational modes in CsSnBr<sub>3</sub> results from electronic rotation–nuclear translation coupling, in analogy to rotation–translation coupling effects in molecular plastic crystals. We also identify electronic fluctuations in the Cs cation that couple to distortions in the surrounding Sn-Br cubic coordination environment. We anticipate that our approach and resulting insights into electronic fluctuations will aid in further understanding the role of the fluctuating lattice in determining important physical properties of halide perovskites and beyond.</p>","PeriodicalId":33,"journal":{"name":"Chemistry of Materials","volume":"36 19","pages":"9442–9459 9442–9459"},"PeriodicalIF":7.0000,"publicationDate":"2024-09-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Dynamic Local Symmetry Fluctuations of Electron Density in Halide Perovskites\",\"authors\":\"Colin M. Hylton-Farrington, and , Richard C. Remsing*, \",\"doi\":\"10.1021/acs.chemmater.4c0112110.1021/acs.chemmater.4c01121\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Metal halide perovskites have emerged as an exciting class of materials for applications in solar energy harvesting, optical devices, catalysis, and other photophysical applications. Many of the exciting properties of halide perovskites are tied to their soft, dynamic, and anharmonic lattice. In particular, the precise coupling between anharmonic lattice dynamics and electronic fluctuations is not completely understood. To build an understanding of this coupling, we use ab initio molecular dynamics simulations supplemented by the calculation of maximally localized Wannier functions to carry out a dynamic group theory analysis of local electron density fluctuations and how these fluctuations are coupled to lattice fluctuations in the model inorganic halide perovskite CsSnBr<sub>3</sub>. We detail symmetry-dependent couplings between vibrational modes including octahedral tilting. Importantly, we suggest that the large anharmonicity of some of the vibrational modes in CsSnBr<sub>3</sub> results from electronic rotation–nuclear translation coupling, in analogy to rotation–translation coupling effects in molecular plastic crystals. We also identify electronic fluctuations in the Cs cation that couple to distortions in the surrounding Sn-Br cubic coordination environment. We anticipate that our approach and resulting insights into electronic fluctuations will aid in further understanding the role of the fluctuating lattice in determining important physical properties of halide perovskites and beyond.</p>\",\"PeriodicalId\":33,\"journal\":{\"name\":\"Chemistry of Materials\",\"volume\":\"36 19\",\"pages\":\"9442–9459 9442–9459\"},\"PeriodicalIF\":7.0000,\"publicationDate\":\"2024-09-18\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Chemistry of Materials\",\"FirstCategoryId\":\"88\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.chemmater.4c01121\",\"RegionNum\":2,\"RegionCategory\":\"材料科学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Chemistry of Materials","FirstCategoryId":"88","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.chemmater.4c01121","RegionNum":2,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
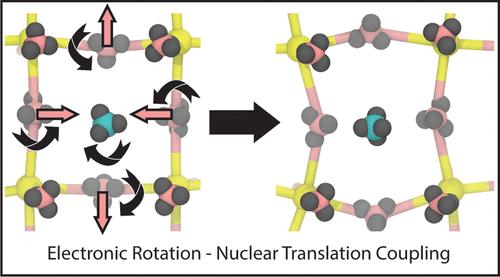
Dynamic Local Symmetry Fluctuations of Electron Density in Halide Perovskites
Metal halide perovskites have emerged as an exciting class of materials for applications in solar energy harvesting, optical devices, catalysis, and other photophysical applications. Many of the exciting properties of halide perovskites are tied to their soft, dynamic, and anharmonic lattice. In particular, the precise coupling between anharmonic lattice dynamics and electronic fluctuations is not completely understood. To build an understanding of this coupling, we use ab initio molecular dynamics simulations supplemented by the calculation of maximally localized Wannier functions to carry out a dynamic group theory analysis of local electron density fluctuations and how these fluctuations are coupled to lattice fluctuations in the model inorganic halide perovskite CsSnBr3. We detail symmetry-dependent couplings between vibrational modes including octahedral tilting. Importantly, we suggest that the large anharmonicity of some of the vibrational modes in CsSnBr3 results from electronic rotation–nuclear translation coupling, in analogy to rotation–translation coupling effects in molecular plastic crystals. We also identify electronic fluctuations in the Cs cation that couple to distortions in the surrounding Sn-Br cubic coordination environment. We anticipate that our approach and resulting insights into electronic fluctuations will aid in further understanding the role of the fluctuating lattice in determining important physical properties of halide perovskites and beyond.
期刊介绍:
The journal Chemistry of Materials focuses on publishing original research at the intersection of materials science and chemistry. The studies published in the journal involve chemistry as a prominent component and explore topics such as the design, synthesis, characterization, processing, understanding, and application of functional or potentially functional materials. The journal covers various areas of interest, including inorganic and organic solid-state chemistry, nanomaterials, biomaterials, thin films and polymers, and composite/hybrid materials. The journal particularly seeks papers that highlight the creation or development of innovative materials with novel optical, electrical, magnetic, catalytic, or mechanical properties. It is essential that manuscripts on these topics have a primary focus on the chemistry of materials and represent a significant advancement compared to prior research. Before external reviews are sought, submitted manuscripts undergo a review process by a minimum of two editors to ensure their appropriateness for the journal and the presence of sufficient evidence of a significant advance that will be of broad interest to the materials chemistry community.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


