利用深度学习生成蛋白质结构以发现通路
IF 5.5
1区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
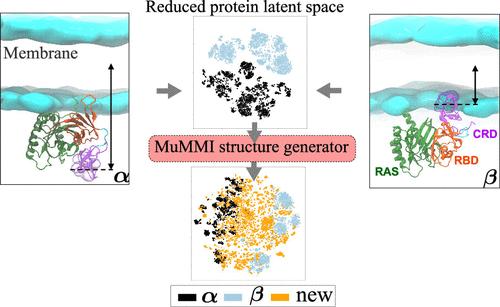
从根本上说,在分子水平上解决生物现象的复杂细节问题,受到实验探究的长度和时间尺度的限制。各种尺度的分子动力学(MD)模拟是一种功能强大的工具,经常被用来提供超出实验分辨率的有价值的生物学见解。然而,虽然在所需的空间分辨率下观察蛋白质等的长期稳定构型相对简单,但模拟这些状态之间更有趣的罕见转变所需的时间往往比当今最大的超级计算机所能处理的时间长几个数量级。这一挑战的一个常见方面是路径发现,在这种情况下,科学现象的起始和结束状态是已知的,或者可以近似得到,但两者之间的机理细节却是未知的。在这里,我们提出了一种基于表征学习的解决方案,利用抽象表征空间中的内插法和外推法合成潜在的过渡状态,并通过 MD 模拟自动进行验证。合成的过渡状态的新模拟随后被纳入表征学习,从而形成了一个目标路径采样的迭代框架。我们的方法通过使用粗粒度 MD 模拟恢复 RAS-RAF 蛋白结构域 (CRD) 从无膜到与膜相互作用的转变过程进行了演示。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Generating Protein Structures for Pathway Discovery Using Deep Learning
Resolving the intricate details of biological phenomena at the molecular level is fundamentally limited by both length- and time scales that can be probed experimentally. Molecular dynamics (MD) simulations at various scales are powerful tools frequently employed to offer valuable biological insights beyond experimental resolution. However, while it is relatively simple to observe long-lived, stable configurations of, for example, proteins, at the required spatial resolution, simulating the more interesting rare transitions between such states often takes orders of magnitude longer than what is feasible even on the largest supercomputers available today. One common aspect of this challenge is pathway discovery, where the start and end states of a scientific phenomenon are known or can be approximated, but the mechanistic details in between are unknown. Here, we propose a representation-learning-based solution that uses interpolation and extrapolation in an abstract representation space to synthesize potential transition states, which are automatically validated using MD simulations. The new simulations of the synthesized transition states are subsequently incorporated into the representation learning, leading to an iterative framework for targeted path sampling. Our approach is demonstrated by recovering the transition of a RAS-RAF protein domain (CRD) from membrane-free to interacting with the membrane using coarse-grain MD simulations.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Journal of Chemical Theory and Computation
化学-物理:原子、分子和化学物理
CiteScore
9.90
自引率
16.40%
发文量
568
审稿时长
1 months
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


