使用线性化对密度函数理论的半经典非绝热分子动力学
IF 5.7
1区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
非绝热分子动力学是模拟电子激发分子非辐射衰变的有效方法。它的准确性在很大程度上取决于势能面的质量,而对于具有充分集合平均的长时间直接动力学模拟来说,它的经济承受能力在很大程度上取决于所需电子结构计算的成本。线性化对密度函数理论(L-PDFT)是最近开发的一种后自洽场多参量方法,它可以对势能面进行建模,其精度与昂贵的多参量扰动理论相似,但计算成本却与基础的多配置自洽场方法相似。在此,我们整合了 SHARC 动力学和 PySCF 电子结构代码,利用 L-PDFT 进行电子非绝热计算,并使用组合程序研究顺式氮杂甲烷的光异构化反应。我们的研究表明,L-PDFT 能够成功模拟光异构化反应而不发生碰撞,其结果类似于成本更高的扩展多态完整活性空间二阶扰动理论。这表明 L-PDFT 可以模拟内部转化,并展示了它在更广泛的光动力学应用中的前景。本文章由计算机程序翻译,如有差异,请以英文原文为准。
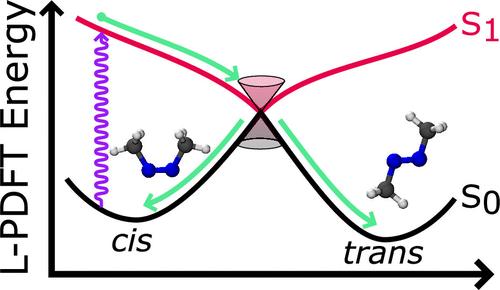
Semiclassical Nonadiabatic Molecular Dynamics Using Linearized Pair-Density Functional Theory
Nonadiabatic molecular dynamics is an effective method for modeling nonradiative decay in electronically excited molecules. Its accuracy depends strongly on the quality of the potential energy surfaces, and its affordability for long direct-dynamic simulations with adequate ensemble averaging depends strongly on the cost of the required electronic structure calculations. Linearized pair-density functional theory (L-PDFT) is a recently developed post-self-consistent-field multireference method that can model potential energy surfaces with an accuracy similar to expensive multireference perturbation theories but at a computational cost similar to the underlying multiconfiguration self-consistent field method. Here, we integrate the SHARC dynamics and PySCF electronic structure code to utilize L-PDFT for electronically nonadiabatic calculations and use the combined programs to study the photoisomerization reaction of cis-azomethane. We show that L-PDFT is able to successfully simulate the photoisomerization without crashes, and it yields results similar to the more expensive extended multistate complete active space second-order perturbation theory. This shows that L-PDFT can model internal conversion, and it demonstrates its promise for broader photodynamics applications.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Journal of Chemical Theory and Computation
化学-物理:原子、分子和化学物理
CiteScore
9.90
自引率
16.40%
发文量
568
审稿时长
1 months
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


