{"title":"聚噻吩的化学可转移电子粗粒化。","authors":"Zheng Yu, Nicholas E Jackson","doi":"10.1021/acs.jctc.4c00804","DOIUrl":null,"url":null,"abstract":"<p><p>Recent advances in machine-learning-based electronic coarse graining (ECG) methods have demonstrated the potential to enable electronic predictions in soft materials at mesoscopic length scales. However, previous ECG models have yet to confront the issue of chemical transferability. In this study, we develop chemically transferable ECG models for polythiophenes using graph neural networks. Our models are trained on a data set that samples over the conformational space of random polythiophene sequences generated with 15 different monomer chemistries and three different degrees of polymerization. We systematically explore the impact of coarse-grained representation on ECG accuracy, highlighting the significance of preserving the C-β coordinates in thiophene. We also find that integrating unique polymer sequences into training enhances the model performance more efficiently than augmenting conformational sampling for sequences already in the training data set. Moreover, our ECG models, developed initially for one property and one level of quantum chemical theory, can be efficiently transferred to related properties and higher levels of theory with minimal additional data. The chemically transferable ECG model introduced in this work will serve as a foundation model for new classes of chemically transferable ECG predictions across chemical space.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":" ","pages":"9116-9127"},"PeriodicalIF":5.7000,"publicationDate":"2024-10-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Chemically Transferable Electronic Coarse Graining for Polythiophenes.\",\"authors\":\"Zheng Yu, Nicholas E Jackson\",\"doi\":\"10.1021/acs.jctc.4c00804\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Recent advances in machine-learning-based electronic coarse graining (ECG) methods have demonstrated the potential to enable electronic predictions in soft materials at mesoscopic length scales. However, previous ECG models have yet to confront the issue of chemical transferability. In this study, we develop chemically transferable ECG models for polythiophenes using graph neural networks. Our models are trained on a data set that samples over the conformational space of random polythiophene sequences generated with 15 different monomer chemistries and three different degrees of polymerization. We systematically explore the impact of coarse-grained representation on ECG accuracy, highlighting the significance of preserving the C-β coordinates in thiophene. We also find that integrating unique polymer sequences into training enhances the model performance more efficiently than augmenting conformational sampling for sequences already in the training data set. Moreover, our ECG models, developed initially for one property and one level of quantum chemical theory, can be efficiently transferred to related properties and higher levels of theory with minimal additional data. The chemically transferable ECG model introduced in this work will serve as a foundation model for new classes of chemically transferable ECG predictions across chemical space.</p>\",\"PeriodicalId\":45,\"journal\":{\"name\":\"Journal of Chemical Theory and Computation\",\"volume\":\" \",\"pages\":\"9116-9127\"},\"PeriodicalIF\":5.7000,\"publicationDate\":\"2024-10-22\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Theory and Computation\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://doi.org/10.1021/acs.jctc.4c00804\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/10/7 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jctc.4c00804","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/10/7 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Chemically Transferable Electronic Coarse Graining for Polythiophenes.
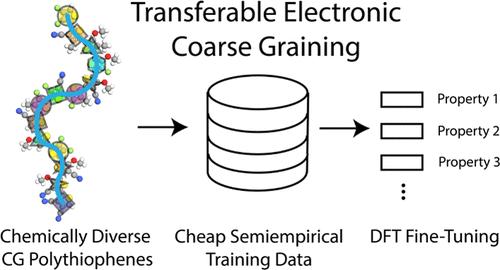
Recent advances in machine-learning-based electronic coarse graining (ECG) methods have demonstrated the potential to enable electronic predictions in soft materials at mesoscopic length scales. However, previous ECG models have yet to confront the issue of chemical transferability. In this study, we develop chemically transferable ECG models for polythiophenes using graph neural networks. Our models are trained on a data set that samples over the conformational space of random polythiophene sequences generated with 15 different monomer chemistries and three different degrees of polymerization. We systematically explore the impact of coarse-grained representation on ECG accuracy, highlighting the significance of preserving the C-β coordinates in thiophene. We also find that integrating unique polymer sequences into training enhances the model performance more efficiently than augmenting conformational sampling for sequences already in the training data set. Moreover, our ECG models, developed initially for one property and one level of quantum chemical theory, can be efficiently transferred to related properties and higher levels of theory with minimal additional data. The chemically transferable ECG model introduced in this work will serve as a foundation model for new classes of chemically transferable ECG predictions across chemical space.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


