Timothy D Loose, Patrick G Sahrmann, Thomas S Qu, Gregory A Voth
{"title":"换一杯马提尼还是会让你宿醉。","authors":"Timothy D Loose, Patrick G Sahrmann, Thomas S Qu, Gregory A Voth","doi":"10.1021/acs.jctc.4c00868","DOIUrl":null,"url":null,"abstract":"<p><p>The Martini 3.0 coarse-grained force field, which was parametrized to better capture transferability in top-down coarse-grained models, is analyzed to assess its accuracy in representing thermodynamic and structural properties with respect to the underlying atomistic representation of the system. These results are compared to those obtained following the principles of statistical mechanics that start from the same underlying atomistic system. To this end, the potentials of mean force for lateral association in Martini 3.0 binary lipid bilayers are decomposed into their entropic and enthalpic components and compared to those of corresponding atomistic bilayers that have been projected onto equivalent coarse-grained mappings but evolved under the fully atomistic forces. This is accomplished by applying the reversible work theorem to lateral pair correlation functions between coarse-grained lipid beads taken at a range of different temperatures. The entropy-enthalpy decompositions provide a metric by which the underlying statistical mechanical properties of Martini can be investigated. Overall, Martini 3.0 is found to fail to properly partition entropy and enthalpy for the PMFs compared to the mapped all-atom results, despite changes made to the force field from the Martini 2.0 version. This outcome points to the fact that the development of more accurate top-down coarse-grained models such as Martini will likely necessitate temperature-dependent terms in the corresponding CG force-field; although necessary, this may not be sufficient to improve Martini. In addition to the entropy-enthalpy decompositions, Martini 3.0 produces an incorrect undulation spectrum, in particular at intermediate length scales of biophysical pertinence.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":" ","pages":"9190-9208"},"PeriodicalIF":5.5000,"publicationDate":"2024-10-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11500708/pdf/","citationCount":"0","resultStr":"{\"title\":\"Changing Your Martini Can Still Give You a Hangover.\",\"authors\":\"Timothy D Loose, Patrick G Sahrmann, Thomas S Qu, Gregory A Voth\",\"doi\":\"10.1021/acs.jctc.4c00868\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>The Martini 3.0 coarse-grained force field, which was parametrized to better capture transferability in top-down coarse-grained models, is analyzed to assess its accuracy in representing thermodynamic and structural properties with respect to the underlying atomistic representation of the system. These results are compared to those obtained following the principles of statistical mechanics that start from the same underlying atomistic system. To this end, the potentials of mean force for lateral association in Martini 3.0 binary lipid bilayers are decomposed into their entropic and enthalpic components and compared to those of corresponding atomistic bilayers that have been projected onto equivalent coarse-grained mappings but evolved under the fully atomistic forces. This is accomplished by applying the reversible work theorem to lateral pair correlation functions between coarse-grained lipid beads taken at a range of different temperatures. The entropy-enthalpy decompositions provide a metric by which the underlying statistical mechanical properties of Martini can be investigated. Overall, Martini 3.0 is found to fail to properly partition entropy and enthalpy for the PMFs compared to the mapped all-atom results, despite changes made to the force field from the Martini 2.0 version. This outcome points to the fact that the development of more accurate top-down coarse-grained models such as Martini will likely necessitate temperature-dependent terms in the corresponding CG force-field; although necessary, this may not be sufficient to improve Martini. In addition to the entropy-enthalpy decompositions, Martini 3.0 produces an incorrect undulation spectrum, in particular at intermediate length scales of biophysical pertinence.</p>\",\"PeriodicalId\":45,\"journal\":{\"name\":\"Journal of Chemical Theory and Computation\",\"volume\":\" \",\"pages\":\"9190-9208\"},\"PeriodicalIF\":5.5000,\"publicationDate\":\"2024-10-22\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11500708/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Theory and Computation\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://doi.org/10.1021/acs.jctc.4c00868\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/10/3 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jctc.4c00868","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/10/3 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
摘要
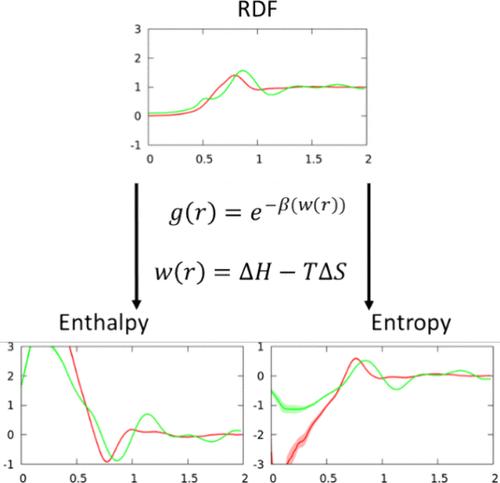
为了更好地捕捉自上而下粗粒度模型的可转移性,对马丁尼 3.0 粗粒度力场进行了参数化分析,以评估其相对于系统的基本原子表征在表示热力学和结构特性方面的准确性。将这些结果与根据统计力学原理(从相同的基础原子系统出发)获得的结果进行比较。为此,我们将 Martini 3.0 二元脂质双分子层横向联合的平均力势分解为熵和焓两部分,并与投影到等效粗粒度映射上但在完全原子力作用下演化的相应原子双分子层的平均力势进行比较。这是通过将可逆功定理应用于在一系列不同温度下提取的粗粒度脂珠之间的侧对相关函数来实现的。熵焓分解提供了一种度量方法,通过它可以研究 Martini 的基本统计机械特性。总体而言,尽管力场与 Martini 2.0 版本相比有所变化,但与映射的全原子结果相比,Martini 3.0 未能正确分配 PMF 的熵和焓。这一结果表明,要开发更精确的自上而下粗粒度模型(如马提尼模型),可能需要在相应的 CG 力场中加入温度相关项;尽管这是必要的,但这可能不足以改善马提尼模型。除了熵焓分解之外,Martini 3.0 还产生了不正确的起伏谱,尤其是在与生物物理相关的中等长度尺度上。
Changing Your Martini Can Still Give You a Hangover.
The Martini 3.0 coarse-grained force field, which was parametrized to better capture transferability in top-down coarse-grained models, is analyzed to assess its accuracy in representing thermodynamic and structural properties with respect to the underlying atomistic representation of the system. These results are compared to those obtained following the principles of statistical mechanics that start from the same underlying atomistic system. To this end, the potentials of mean force for lateral association in Martini 3.0 binary lipid bilayers are decomposed into their entropic and enthalpic components and compared to those of corresponding atomistic bilayers that have been projected onto equivalent coarse-grained mappings but evolved under the fully atomistic forces. This is accomplished by applying the reversible work theorem to lateral pair correlation functions between coarse-grained lipid beads taken at a range of different temperatures. The entropy-enthalpy decompositions provide a metric by which the underlying statistical mechanical properties of Martini can be investigated. Overall, Martini 3.0 is found to fail to properly partition entropy and enthalpy for the PMFs compared to the mapped all-atom results, despite changes made to the force field from the Martini 2.0 version. This outcome points to the fact that the development of more accurate top-down coarse-grained models such as Martini will likely necessitate temperature-dependent terms in the corresponding CG force-field; although necessary, this may not be sufficient to improve Martini. In addition to the entropy-enthalpy decompositions, Martini 3.0 produces an incorrect undulation spectrum, in particular at intermediate length scales of biophysical pertinence.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


