用于激发态计算的子空间搜索量子虚时间演化
IF 5.5
1区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
随着噪声中量子(NISQ)器件的出现,激发态量子系统正引起人们的极大兴趣。虽然小分子系统的基态通常使用混合变分算法(如变分量子求解器(VQE))进行探索,但激发态的研究受到的关注要少得多,部分原因是缺乏高效算法。在这项工作中,我们介绍了子空间搜索量子虚时间演化(SSQITE)方法,该方法通过整合子空间搜索变分量子特征分解器(SSVQE)和变分量子虚时间演化(VarQITE)方法的关键要素,利用量子器件计算激发态。通过计算基准模型系统(包括 H2 和 LiH 分子)的低洼激发态,证明了 SSQITE 的有效性。此外,还使用了一个玩具哈密顿,以证明 VarQITE 在避免局部极小值方面的稳健性可扩展到其在激发态算法中的应用。凭借在避免局部极小值方面的稳健性,SSQITE 显示出在广泛应用中推进激发态量子计算的前景。本文章由计算机程序翻译,如有差异,请以英文原文为准。
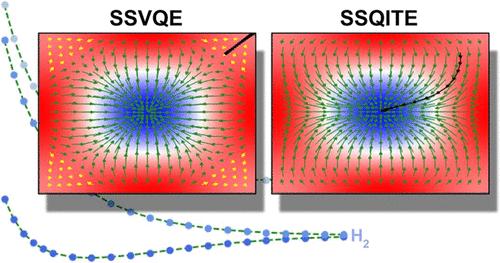
Subspace-Search Quantum Imaginary Time Evolution for Excited State Computations
Quantum systems in excited states are attracting significant interest with the advent of noisy intermediate-scale quantum (NISQ) devices. While ground states of small molecular systems are typically explored using hybrid variational algorithms like the variational quantum eigensolver (VQE), the study of excited states has received much less attention, partly due to the absence of efficient algorithms. In this work, we introduce the subspace search quantum imaginary time evolution (SSQITE) method, which calculates excited states using quantum devices by integrating key elements of the subspace search variational quantum eigensolver (SSVQE) and the variational quantum imaginary time evolution (VarQITE) method. The effectiveness of SSQITE is demonstrated through calculations of low-lying excited states of benchmark model systems including H2 and LiH molecules. A toy Hamiltonian is also employed to demonstrate that the robustness of VarQITE in avoiding local minima extends to its use in excited state algorithms. With this robustness in avoiding local minima, SSQITE shows promise for advancing quantum computations of excited states across a wide range of applications.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Journal of Chemical Theory and Computation
化学-物理:原子、分子和化学物理
CiteScore
9.90
自引率
16.40%
发文量
568
审稿时长
1 months
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


