实现一氧化碳的精确 Ab Initio 基底势能和电偶极矩函数
IF 5.5
1区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
利用单参量耦合簇方法(达到 CCSDTQP 近似水平),结合增强型核价相关一致基集 aug-cc-pCVnZ(达到 octuple-zeta 质量),获得了一氧化碳分子在其基态电子 X1Σ+ 中的精确势能和电偶极矩函数。讨论了标量相对论效应、绝热效应和非绝热效应。将 ab initio 预测函数与实验得出的对应函数进行了比较。本文章由计算机程序翻译,如有差异,请以英文原文为准。
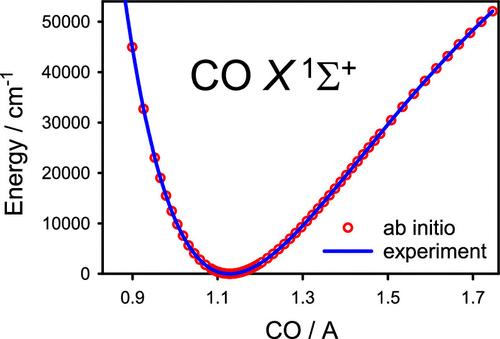
Toward Accurate Ab Initio Ground-State Potential Energy and Electric Dipole Moment Functions of Carbon Monoxide
Accurate potential energy and electric dipole moment functions of the CO molecule in its ground electronic state X1Σ+ have been obtained using the single-reference coupled-cluster approach, up to the CCSDTQP level of approximation, in conjunction with the augmented core–valence correlation-consistent basis sets, aug-cc-pCVnZ, up to octuple-zeta quality. The scalar relativistic, adiabatic, and nonadiabatic effects were discussed. The ab initio predicted functions were compared with their experimentally derived counterparts.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Journal of Chemical Theory and Computation
化学-物理:原子、分子和化学物理
CiteScore
9.90
自引率
16.40%
发文量
568
审稿时长
1 months
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


