四体非绝热指数波函数的相对论修正
IF 5.7
1区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
我们提出了一种计算氢分子相对论修正的方法,其精确度大大超过了以往所有的文献结果。该方法利用了显式相关非绝热指数波函数,从而等效地处理了电子和原子核。所提出的方法可应用于任何振动态,包括高度激发态。相对论修正的数值精度达到几千赫兹(∼10-7 cm-1),低于最佳实验精度。本文章由计算机程序翻译,如有差异,请以英文原文为准。
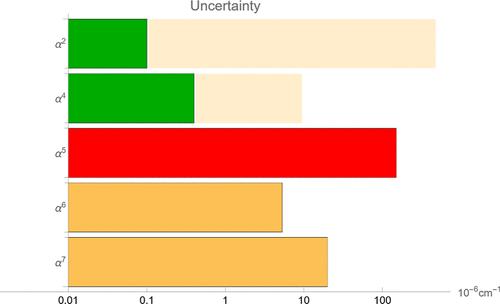
Relativistic Correction from the Four-Body Nonadiabatic Exponential Wave Function
We present a method for calculating the relativistic correction in hydrogen molecules that significantly exceeds the accuracy of all the previous literature results. This method utilizes the explicitly correlated nonadiabatic exponential wave function, and thus treats electrons and nuclei equivalently. The proposed method can be applied to any rovibrational state, including highly excited ones. The numerical precision of the relativistic correction reaches several kHz (∼10–7 cm–1), which is below the best experimental accuracy.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Journal of Chemical Theory and Computation
化学-物理:原子、分子和化学物理
CiteScore
9.90
自引率
16.40%
发文量
568
审稿时长
1 months
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


