识别影响单细胞数据中细胞状态丰度的基因变异
IF 31.7
1区 生物学
Q1 GENETICS & HEREDITY
引用次数: 0
摘要
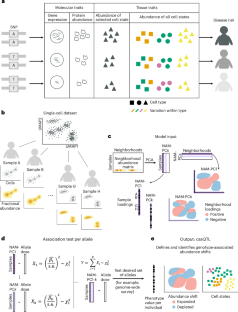
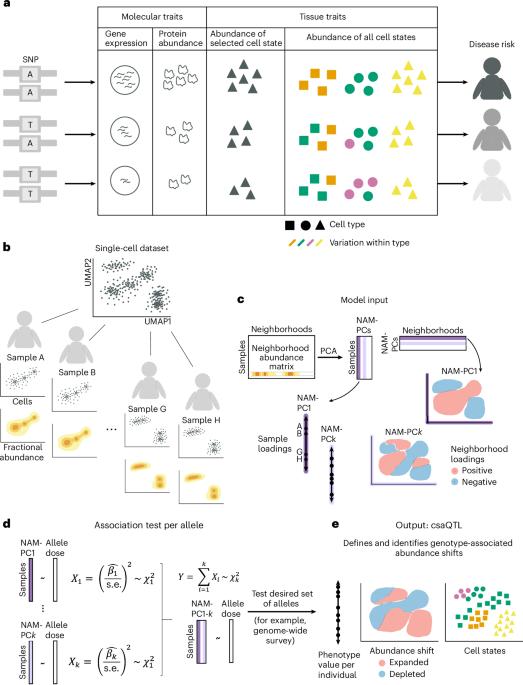
疾病风险等位基因会影响体内细胞的组成,但对单细胞图谱揭示的细胞状态的遗传效应建模却很困难,因为变异相关状态可能反映了图谱细胞特征的不同组合,而这些组合很难预先确定。我们介绍了基因型-邻近关联(GeNA),这是一种在高维单细胞数据集中识别细胞状态丰度定量性状位点(csaQTLs)的统计工具。GeNA 不测试与预定义细胞状态的关联,而是灵活地确定其丰度与遗传变异关联最大的细胞状态。在对 969 人的外周血单细胞 RNA 测序分析进行的全基因组调查中,GeNA 发现了五个与免疫细胞状态相对丰度变化相关的独立位点。例如,rs3003-T(P = 1.96 × 10-11)与表达肿瘤坏死因子反应程序的自然杀伤细胞数量增加有关。这种 csaQTL 与银屑病风险的增加有关,银屑病是一种自身免疫性疾病,对抗肿瘤坏死因子治疗有反应。灵活表征颗粒细胞状态的 csaQTL 可能有助于阐明遗传背景如何改变细胞组成,从而导致疾病风险。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Identifying genetic variants that influence the abundance of cell states in single-cell data
Disease risk alleles influence the composition of cells present in the body, but modeling genetic effects on the cell states revealed by single-cell profiling is difficult because variant-associated states may reflect diverse combinations of the profiled cell features that are challenging to predefine. We introduce Genotype–Neighborhood Associations (GeNA), a statistical tool to identify cell-state abundance quantitative trait loci (csaQTLs) in high-dimensional single-cell datasets. Instead of testing associations to predefined cell states, GeNA flexibly identifies the cell states whose abundance is most associated with genetic variants. In a genome-wide survey of single-cell RNA sequencing peripheral blood profiling from 969 individuals, GeNA identifies five independent loci associated with shifts in the relative abundance of immune cell states. For example, rs3003-T (P = 1.96 × 10−11) associates with increased abundance of natural killer cells expressing tumor necrosis factor response programs. This csaQTL colocalizes with increased risk for psoriasis, an autoimmune disease that responds to anti-tumor necrosis factor treatments. Flexibly characterizing csaQTLs for granular cell states may help illuminate how genetic background alters cellular composition to confer disease risk. GeNA identifies cell-state abundance quantitative trait loci (csaQTLs) in single-cell RNA sequencing data. Applied to OneK1K, GeNA identifies natural killer cell and myeloid csaQTLs and implicates interferon-α-related cell states using a polygenic risk score for systemic lupus erythematosus.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Nature genetics
生物-遗传学
CiteScore
43.00
自引率
2.60%
发文量
241
审稿时长
3 months
期刊介绍:
Nature Genetics publishes the very highest quality research in genetics. It encompasses genetic and functional genomic studies on human and plant traits and on other model organisms. Current emphasis is on the genetic basis for common and complex diseases and on the functional mechanism, architecture and evolution of gene networks, studied by experimental perturbation.
Integrative genetic topics comprise, but are not limited to:
-Genes in the pathology of human disease
-Molecular analysis of simple and complex genetic traits
-Cancer genetics
-Agricultural genomics
-Developmental genetics
-Regulatory variation in gene expression
-Strategies and technologies for extracting function from genomic data
-Pharmacological genomics
-Genome evolution
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


