{"title":"无显式非绝热耦合的机器学习辅助量子-经典混合动力学:过硝酸光解离的应用","authors":"Mahesh K. Sit, Subhasish Das and Kousik Samanta*, ","doi":"10.1021/acs.jpca.4c0287610.1021/acs.jpca.4c02876","DOIUrl":null,"url":null,"abstract":"<p >We have devised a hybrid quantum–classical scheme utilizing machine-learned potential energy surfaces (PES), which circumvents the need for explicit computation of nonadiabatic coupling elements. The quantities necessary to account for the nonadiabatic effects are directly obtained from the PESs. The simulation of dynamics is based on the fewest-switches surface-hopping method. We applied this scheme to model the photodissociation of both N–O and O–O bonds in a conformer of peroxynitric acid (HO<sub>2</sub>NO<sub>2</sub>). Adiabatic PES data for the six lowest states of this molecule were computed at the CASSCF level for various nuclear configurations. These served as the training data for the machine-learning models for the PESs. The dynamics simulation was initiated on the lowest optically bright singlet excited state (S<sub>4</sub>) and propagated along the two Jacobi coordinates <i></i><math><msub><mrow><mover><mi>J</mi><mo>→</mo></mover></mrow><mrow><mn>1</mn></mrow></msub></math> and <i></i><math><msub><mrow><mover><mi>J</mi><mo>→</mo></mover></mrow><mrow><mn>2</mn></mrow></msub></math> while accounting for the nonadiabatic effects through transitions between PESs. Our analysis revealed that there is a very high chance of dissociation of the N–O bond leading to the HO<sub>2</sub> and NO<sub>2</sub> fragments.</p>","PeriodicalId":59,"journal":{"name":"The Journal of Physical Chemistry A","volume":"128 38","pages":"8244–8253 8244–8253"},"PeriodicalIF":2.8000,"publicationDate":"2024-09-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Machine Learning-Assisted Mixed Quantum-Classical Dynamics without Explicit Nonadiabatic Coupling: Application to the Photodissociation of Peroxynitric Acid\",\"authors\":\"Mahesh K. Sit, Subhasish Das and Kousik Samanta*, \",\"doi\":\"10.1021/acs.jpca.4c0287610.1021/acs.jpca.4c02876\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >We have devised a hybrid quantum–classical scheme utilizing machine-learned potential energy surfaces (PES), which circumvents the need for explicit computation of nonadiabatic coupling elements. The quantities necessary to account for the nonadiabatic effects are directly obtained from the PESs. The simulation of dynamics is based on the fewest-switches surface-hopping method. We applied this scheme to model the photodissociation of both N–O and O–O bonds in a conformer of peroxynitric acid (HO<sub>2</sub>NO<sub>2</sub>). Adiabatic PES data for the six lowest states of this molecule were computed at the CASSCF level for various nuclear configurations. These served as the training data for the machine-learning models for the PESs. The dynamics simulation was initiated on the lowest optically bright singlet excited state (S<sub>4</sub>) and propagated along the two Jacobi coordinates <i></i><math><msub><mrow><mover><mi>J</mi><mo>→</mo></mover></mrow><mrow><mn>1</mn></mrow></msub></math> and <i></i><math><msub><mrow><mover><mi>J</mi><mo>→</mo></mover></mrow><mrow><mn>2</mn></mrow></msub></math> while accounting for the nonadiabatic effects through transitions between PESs. Our analysis revealed that there is a very high chance of dissociation of the N–O bond leading to the HO<sub>2</sub> and NO<sub>2</sub> fragments.</p>\",\"PeriodicalId\":59,\"journal\":{\"name\":\"The Journal of Physical Chemistry A\",\"volume\":\"128 38\",\"pages\":\"8244–8253 8244–8253\"},\"PeriodicalIF\":2.8000,\"publicationDate\":\"2024-09-16\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"The Journal of Physical Chemistry A\",\"FirstCategoryId\":\"1\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jpca.4c02876\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry A","FirstCategoryId":"1","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jpca.4c02876","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
摘要
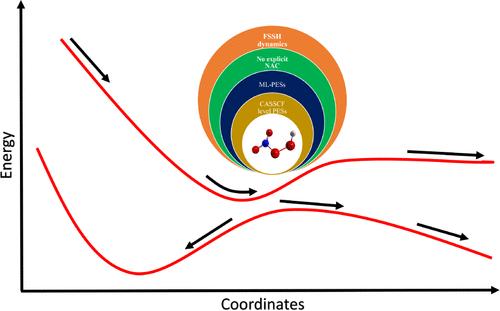
我们设计了一种量子-经典混合方案,利用机器学习的势能面(PES),避免了非绝热耦合元素的显式计算。考虑非绝热效应所需的量可以直接从势能面中获得。动力学模拟基于最少开关表面跳转法。我们将此方案用于模拟过硝酸(HO2NO2)构象中 N-O 和 O-O 键的光解离。我们在 CASSCF 水平上计算了该分子在不同核构型下六个最低态的绝热 PES 数据。这些数据可作为 PES 机器学习模型的训练数据。动力学模拟从最低的光亮单激发态(S4)开始,沿着两个雅可比坐标 J→1 和 J→2 传播,同时通过 PES 之间的转换考虑非绝热效应。我们的分析表明,N-O 键解离导致产生 HO2 和 NO2 片段的几率非常高。
Machine Learning-Assisted Mixed Quantum-Classical Dynamics without Explicit Nonadiabatic Coupling: Application to the Photodissociation of Peroxynitric Acid
We have devised a hybrid quantum–classical scheme utilizing machine-learned potential energy surfaces (PES), which circumvents the need for explicit computation of nonadiabatic coupling elements. The quantities necessary to account for the nonadiabatic effects are directly obtained from the PESs. The simulation of dynamics is based on the fewest-switches surface-hopping method. We applied this scheme to model the photodissociation of both N–O and O–O bonds in a conformer of peroxynitric acid (HO2NO2). Adiabatic PES data for the six lowest states of this molecule were computed at the CASSCF level for various nuclear configurations. These served as the training data for the machine-learning models for the PESs. The dynamics simulation was initiated on the lowest optically bright singlet excited state (S4) and propagated along the two Jacobi coordinates and while accounting for the nonadiabatic effects through transitions between PESs. Our analysis revealed that there is a very high chance of dissociation of the N–O bond leading to the HO2 and NO2 fragments.
期刊介绍:
The Journal of Physical Chemistry A is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


