英国生物库中基因组、外显子组和估算的遗传关联信号的产量
IF 31.7
1区 生物学
Q1 GENETICS & HEREDITY
引用次数: 0
摘要
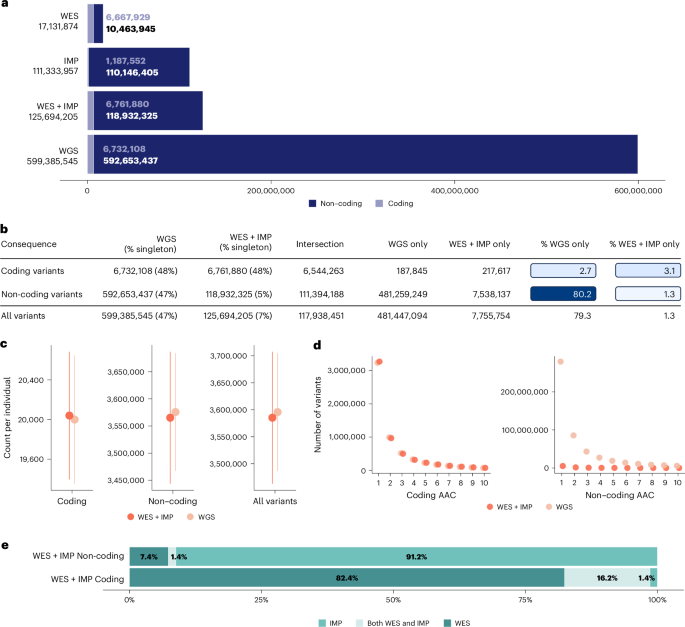
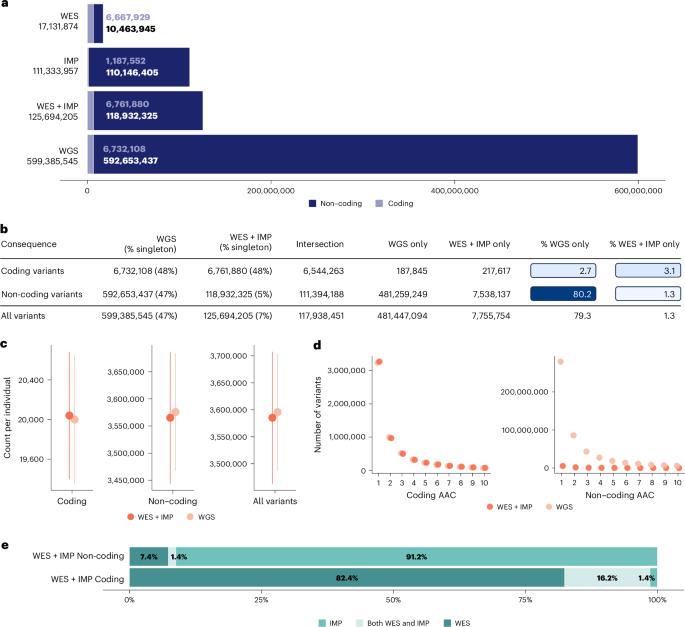
全基因组测序(WGS)、全外显子组测序(WES)和带归因的阵列基因分型(IMP)是评估遗传变异及其与医学相关表型关联的常用策略。迄今为止,这些方法在应用于数十万样本以发现复杂性状遗传信号时的收益还没有系统的实证评估。利用英国生物库中 149195 个个体的 100 个复杂性状的数据,我们系统地比较了这些策略在遗传关联研究中的相对收益。我们发现,WGS 和 WES 结合阵列和归因(WES + IMP)的关联收益最大。虽然与 WES + IMP 相比,WGS 使检测变体总数增加了约五倍,但在单变体和基于基因的关联分析中,检测到的信号数量仅相差 1%。鉴于 WES + IMP 通常可以节省每个样本的实验室和计算时间及资源,我们评估了将 WES + IMP 应用于更大样本的潜在优势。当我们将 WES + IMP 分析扩展到 468,169 个英国生物库个体时,我们发现随着样本量增加三倍,关联信号增加了约四倍。我们的结论是,优先考虑 WES + IMP 和大样本量,而不是当代的短读数 WGS,将最大限度地增加遗传关联研究的发现数量。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Yield of genetic association signals from genomes, exomes and imputation in the UK Biobank
Whole-genome sequencing (WGS), whole-exome sequencing (WES) and array genotyping with imputation (IMP) are common strategies for assessing genetic variation and its association with medically relevant phenotypes. To date, there has been no systematic empirical assessment of the yield of these approaches when applied to hundreds of thousands of samples to enable the discovery of complex trait genetic signals. Using data for 100 complex traits from 149,195 individuals in the UK Biobank, we systematically compare the relative yield of these strategies in genetic association studies. We find that WGS and WES combined with arrays and imputation (WES + IMP) have the largest association yield. Although WGS results in an approximately fivefold increase in the total number of assayed variants over WES + IMP, the number of detected signals differed by only 1% for both single-variant and gene-based association analyses. Given that WES + IMP typically results in savings of lab and computational time and resources expended per sample, we evaluate the potential benefits of applying WES + IMP to larger samples. When we extend our WES + IMP analyses to 468,169 UK Biobank individuals, we observe an approximately fourfold increase in association signals with the threefold increase in sample size. We conclude that prioritizing WES + IMP and large sample sizes rather than contemporary short-read WGS alternatives will maximize the number of discoveries in genetic association studies. Comparison of association signals in UK Biobank using different strategies for assessing genetic variation shows that whole-exome sequencing combined with array genotyping and imputation offers similar performance to whole-genome sequencing at a reduced cost.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Nature genetics
生物-遗传学
CiteScore
43.00
自引率
2.60%
发文量
241
审稿时长
3 months
期刊介绍:
Nature Genetics publishes the very highest quality research in genetics. It encompasses genetic and functional genomic studies on human and plant traits and on other model organisms. Current emphasis is on the genetic basis for common and complex diseases and on the functional mechanism, architecture and evolution of gene networks, studied by experimental perturbation.
Integrative genetic topics comprise, but are not limited to:
-Genes in the pathology of human disease
-Molecular analysis of simple and complex genetic traits
-Cancer genetics
-Agricultural genomics
-Developmental genetics
-Regulatory variation in gene expression
-Strategies and technologies for extracting function from genomic data
-Pharmacological genomics
-Genome evolution
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


