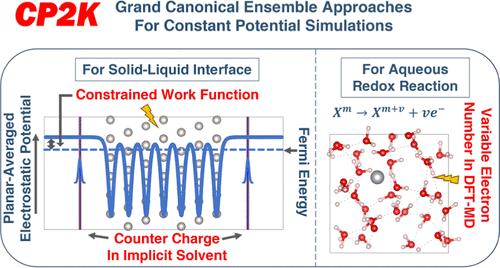
{"title":"用 CP2K 中的大典型集合方法模拟恒定电极电位下的电化学","authors":"Ziwei Chai*, and , Sandra Luber*, ","doi":"10.1021/acs.jctc.4c0067110.1021/acs.jctc.4c00671","DOIUrl":null,"url":null,"abstract":"<p >In electrochemical experiments, the number of electrons of the electrode immersed in the electrolyte is usually variable. Additionally, the numbers of adsorbed substances on the surface of the electrode, the solvent molecules, and counter charge ions in the near-surface region can also vary. Treating electrochemical solid–liquid interfaces with the typical fixed electron number density functional theory (DFT) approach tends to be a challenge. This can be addressed by using grand canonical ensemble approaches. We present the implementation of two grand canonical ensemble approaches in the open-source computational chemistry software CP2K that go beyond the existing canonical ensemble paradigm. The first approach is based on implicit solvent models and explicit atomistic solute (electrode with/without adsorbed species) models, and includes two recent developments: (a) grand canonical self-consistent field (GC-SCF) method (<i>J. Chem. Phys.</i> <b>2017,</b> <i>146,</i> 114104) allowing the electron number of the system to fluctuate naturally and accordingly with the experimental electrode potential, (b) planar counter charge (<i>J. Chem. Phys.</i> <b>2019,</b> <i>150,</i> 041722, <i>Phys. Rev. B</i> <b>2003,</b> <i>68,</i> 245416) salt model completely screening the net charge of the electrode model. In contrast with previous studies, in our implementation, the work function (WF) (absolute electrode potential if the potential drop at the electrolyte–vacuum interface is omitted) is the constrained quantity during an SCF optimization instead of the Fermi energy. The chemical potential of electrons (negative WF) is a natural variable of the grand potential in the GC ensemble of electronic states, and this method can easily achieve stable SCF convergence and obtain an electronic structure that precisely corresponds to a user-specified WF. The second approach referred to as the GC DFT molecular dynamics (DFT-MD) simulation scheme (<i>Phys. Rev. Lett.</i> <b>2002,</b> <i>88,</i> 213002, <i>J. Chem. Phys.</i> <b>2005,</b> <i>122,</i> 234505, <i>J. Am. Chem. Soc.</i> <b>2004,</b> <i>126</i> (12)<i>,</i> 3928–3938) is based on fully explicit modeling the solvent molecules and the ions and is used to calculate the electron chemical potential corresponding to an equilibrium electrochemical half-reaction (<i>M</i><sup>(<i>n</i>+<i>m</i>)+</sup> + <i>ne</i><sup>–</sup> ⇌ <i>M</i><sup><i>m</i>+</sup>) which involves DFT-MD, by allowing the number of electrons to vary during the DFT-MD simulation process. This opens the way for forefront electrochemical calculations in CP2K for a broad range of systems.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":"20 18","pages":"8214–8228 8214–8228"},"PeriodicalIF":5.5000,"publicationDate":"2024-09-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Grand Canonical Ensemble Approaches in CP2K for Modeling Electrochemistry at Constant Electrode Potentials\",\"authors\":\"Ziwei Chai*, and , Sandra Luber*, \",\"doi\":\"10.1021/acs.jctc.4c0067110.1021/acs.jctc.4c00671\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >In electrochemical experiments, the number of electrons of the electrode immersed in the electrolyte is usually variable. Additionally, the numbers of adsorbed substances on the surface of the electrode, the solvent molecules, and counter charge ions in the near-surface region can also vary. Treating electrochemical solid–liquid interfaces with the typical fixed electron number density functional theory (DFT) approach tends to be a challenge. This can be addressed by using grand canonical ensemble approaches. We present the implementation of two grand canonical ensemble approaches in the open-source computational chemistry software CP2K that go beyond the existing canonical ensemble paradigm. The first approach is based on implicit solvent models and explicit atomistic solute (electrode with/without adsorbed species) models, and includes two recent developments: (a) grand canonical self-consistent field (GC-SCF) method (<i>J. Chem. Phys.</i> <b>2017,</b> <i>146,</i> 114104) allowing the electron number of the system to fluctuate naturally and accordingly with the experimental electrode potential, (b) planar counter charge (<i>J. Chem. Phys.</i> <b>2019,</b> <i>150,</i> 041722, <i>Phys. Rev. B</i> <b>2003,</b> <i>68,</i> 245416) salt model completely screening the net charge of the electrode model. In contrast with previous studies, in our implementation, the work function (WF) (absolute electrode potential if the potential drop at the electrolyte–vacuum interface is omitted) is the constrained quantity during an SCF optimization instead of the Fermi energy. The chemical potential of electrons (negative WF) is a natural variable of the grand potential in the GC ensemble of electronic states, and this method can easily achieve stable SCF convergence and obtain an electronic structure that precisely corresponds to a user-specified WF. The second approach referred to as the GC DFT molecular dynamics (DFT-MD) simulation scheme (<i>Phys. Rev. Lett.</i> <b>2002,</b> <i>88,</i> 213002, <i>J. Chem. Phys.</i> <b>2005,</b> <i>122,</i> 234505, <i>J. Am. Chem. Soc.</i> <b>2004,</b> <i>126</i> (12)<i>,</i> 3928–3938) is based on fully explicit modeling the solvent molecules and the ions and is used to calculate the electron chemical potential corresponding to an equilibrium electrochemical half-reaction (<i>M</i><sup>(<i>n</i>+<i>m</i>)+</sup> + <i>ne</i><sup>–</sup> ⇌ <i>M</i><sup><i>m</i>+</sup>) which involves DFT-MD, by allowing the number of electrons to vary during the DFT-MD simulation process. This opens the way for forefront electrochemical calculations in CP2K for a broad range of systems.</p>\",\"PeriodicalId\":45,\"journal\":{\"name\":\"Journal of Chemical Theory and Computation\",\"volume\":\"20 18\",\"pages\":\"8214–8228 8214–8228\"},\"PeriodicalIF\":5.5000,\"publicationDate\":\"2024-09-06\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Theory and Computation\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jctc.4c00671\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jctc.4c00671","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Grand Canonical Ensemble Approaches in CP2K for Modeling Electrochemistry at Constant Electrode Potentials
In electrochemical experiments, the number of electrons of the electrode immersed in the electrolyte is usually variable. Additionally, the numbers of adsorbed substances on the surface of the electrode, the solvent molecules, and counter charge ions in the near-surface region can also vary. Treating electrochemical solid–liquid interfaces with the typical fixed electron number density functional theory (DFT) approach tends to be a challenge. This can be addressed by using grand canonical ensemble approaches. We present the implementation of two grand canonical ensemble approaches in the open-source computational chemistry software CP2K that go beyond the existing canonical ensemble paradigm. The first approach is based on implicit solvent models and explicit atomistic solute (electrode with/without adsorbed species) models, and includes two recent developments: (a) grand canonical self-consistent field (GC-SCF) method (J. Chem. Phys.2017,146, 114104) allowing the electron number of the system to fluctuate naturally and accordingly with the experimental electrode potential, (b) planar counter charge (J. Chem. Phys.2019,150, 041722, Phys. Rev. B2003,68, 245416) salt model completely screening the net charge of the electrode model. In contrast with previous studies, in our implementation, the work function (WF) (absolute electrode potential if the potential drop at the electrolyte–vacuum interface is omitted) is the constrained quantity during an SCF optimization instead of the Fermi energy. The chemical potential of electrons (negative WF) is a natural variable of the grand potential in the GC ensemble of electronic states, and this method can easily achieve stable SCF convergence and obtain an electronic structure that precisely corresponds to a user-specified WF. The second approach referred to as the GC DFT molecular dynamics (DFT-MD) simulation scheme (Phys. Rev. Lett.2002,88, 213002, J. Chem. Phys.2005,122, 234505, J. Am. Chem. Soc.2004,126 (12), 3928–3938) is based on fully explicit modeling the solvent molecules and the ions and is used to calculate the electron chemical potential corresponding to an equilibrium electrochemical half-reaction (M(n+m)+ + ne– ⇌ Mm+) which involves DFT-MD, by allowing the number of electrons to vary during the DFT-MD simulation process. This opens the way for forefront electrochemical calculations in CP2K for a broad range of systems.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


