Yufen Yao, Runduo Liu, Wenchao Li, Wanyi Huang, Yijun Lai, Hai-Bin Luo and Zhe Li*,
{"title":"收敛自适应往返法实现了快速准确的 FEP 计算","authors":"Yufen Yao, Runduo Liu, Wenchao Li, Wanyi Huang, Yijun Lai, Hai-Bin Luo and Zhe Li*, ","doi":"10.1021/acs.jctc.4c0093910.1021/acs.jctc.4c00939","DOIUrl":null,"url":null,"abstract":"<p >The free energy perturbation (FEP) method is a powerful technique for accurate binding free energy calculations, which is crucial for identifying potent ligands with a high affinity in drug discovery. However, the widespread application of FEP is limited by the high computational cost required to achieve equilibrium sampling and the challenges in obtaining converged predictions. In this study, we present the convergence-adaptive roundtrip (CAR) method, which is an enhanced adaptive sampling approach, to address the key challenges in FEP calculations, including the precision-efficiency tradeoff, sampling efficiency, and convergence assessment. By employing on-the-fly convergence analysis to automatically adjust simulation times, enabling efficient traversal of the important phase space through rapid propagation of conformations between different states and eliminating the need for multiple parallel simulations, the CAR method increases convergence and minimizes computational overhead while maintaining calculation accuracy. The performance of the CAR method was evaluated through relative binding free energy (RBFE) calculations on benchmarks comprising four diverse protein–ligand systems. The results demonstrated a significant speedup of over 8-fold compared to conventional FEP methods while maintaining high accuracy. The overall <i>R</i><sup><i>2</i></sup> values of 0.65 and 0.56 were obtained using the combined-structure FEP approach and the single-step FEP approach, respectively, in conjunction with the CAR method. In-depth case studies further highlighted the superior performance of the CAR method in terms of convergence acceleration, improved predicted correlations, and reduced computational costs. The advancement of the CAR method makes it a highly effective approach, enhancing the applicability of FEP in drug discovery.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":"20 18","pages":"8354–8366 8354–8366"},"PeriodicalIF":5.5000,"publicationDate":"2024-09-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Convergence-Adaptive Roundtrip Method Enables Rapid and Accurate FEP Calculations\",\"authors\":\"Yufen Yao, Runduo Liu, Wenchao Li, Wanyi Huang, Yijun Lai, Hai-Bin Luo and Zhe Li*, \",\"doi\":\"10.1021/acs.jctc.4c0093910.1021/acs.jctc.4c00939\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >The free energy perturbation (FEP) method is a powerful technique for accurate binding free energy calculations, which is crucial for identifying potent ligands with a high affinity in drug discovery. However, the widespread application of FEP is limited by the high computational cost required to achieve equilibrium sampling and the challenges in obtaining converged predictions. In this study, we present the convergence-adaptive roundtrip (CAR) method, which is an enhanced adaptive sampling approach, to address the key challenges in FEP calculations, including the precision-efficiency tradeoff, sampling efficiency, and convergence assessment. By employing on-the-fly convergence analysis to automatically adjust simulation times, enabling efficient traversal of the important phase space through rapid propagation of conformations between different states and eliminating the need for multiple parallel simulations, the CAR method increases convergence and minimizes computational overhead while maintaining calculation accuracy. The performance of the CAR method was evaluated through relative binding free energy (RBFE) calculations on benchmarks comprising four diverse protein–ligand systems. The results demonstrated a significant speedup of over 8-fold compared to conventional FEP methods while maintaining high accuracy. The overall <i>R</i><sup><i>2</i></sup> values of 0.65 and 0.56 were obtained using the combined-structure FEP approach and the single-step FEP approach, respectively, in conjunction with the CAR method. In-depth case studies further highlighted the superior performance of the CAR method in terms of convergence acceleration, improved predicted correlations, and reduced computational costs. The advancement of the CAR method makes it a highly effective approach, enhancing the applicability of FEP in drug discovery.</p>\",\"PeriodicalId\":45,\"journal\":{\"name\":\"Journal of Chemical Theory and Computation\",\"volume\":\"20 18\",\"pages\":\"8354–8366 8354–8366\"},\"PeriodicalIF\":5.5000,\"publicationDate\":\"2024-09-05\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Theory and Computation\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jctc.4c00939\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jctc.4c00939","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
摘要
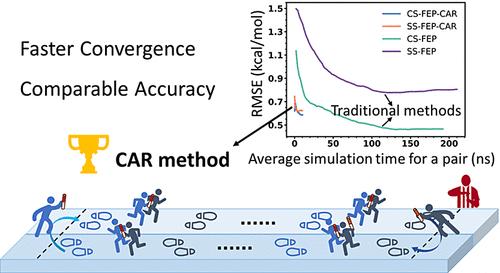
自由能扰动(FEP)方法是一种用于精确计算结合自由能的强大技术,对于在药物发现过程中确定具有高亲和力的强效配体至关重要。然而,由于实现平衡采样所需的计算成本较高,以及获得收敛预测的挑战,FEP 的广泛应用受到了限制。在本研究中,我们提出了收敛自适应往返(CAR)方法,这是一种增强型自适应采样方法,可解决 FEP 计算中的关键难题,包括精度-效率权衡、采样效率和收敛性评估。CAR 方法采用即时收敛分析自动调整模拟时间,通过在不同状态之间快速传播构象实现对重要相空间的高效遍历,无需进行多次并行模拟,从而在保持计算精度的同时提高了收敛性并最大限度地减少了计算开销。通过对四种不同蛋白质配体系统的基准进行相对结合自由能(RBFE)计算,对 CAR 方法的性能进行了评估。结果表明,与传统的 FEP 方法相比,在保持高精度的同时,计算速度明显加快了 8 倍以上。结合 CAR 方法使用组合结构 FEP 方法和单步 FEP 方法得到的总体 R2 值分别为 0.65 和 0.56。深入的案例研究进一步凸显了 CAR 方法在加速收敛、提高预测相关性和降低计算成本方面的优越性能。CAR 方法的先进性使其成为一种高效的方法,提高了 FEP 在药物发现中的适用性。
Convergence-Adaptive Roundtrip Method Enables Rapid and Accurate FEP Calculations
The free energy perturbation (FEP) method is a powerful technique for accurate binding free energy calculations, which is crucial for identifying potent ligands with a high affinity in drug discovery. However, the widespread application of FEP is limited by the high computational cost required to achieve equilibrium sampling and the challenges in obtaining converged predictions. In this study, we present the convergence-adaptive roundtrip (CAR) method, which is an enhanced adaptive sampling approach, to address the key challenges in FEP calculations, including the precision-efficiency tradeoff, sampling efficiency, and convergence assessment. By employing on-the-fly convergence analysis to automatically adjust simulation times, enabling efficient traversal of the important phase space through rapid propagation of conformations between different states and eliminating the need for multiple parallel simulations, the CAR method increases convergence and minimizes computational overhead while maintaining calculation accuracy. The performance of the CAR method was evaluated through relative binding free energy (RBFE) calculations on benchmarks comprising four diverse protein–ligand systems. The results demonstrated a significant speedup of over 8-fold compared to conventional FEP methods while maintaining high accuracy. The overall R2 values of 0.65 and 0.56 were obtained using the combined-structure FEP approach and the single-step FEP approach, respectively, in conjunction with the CAR method. In-depth case studies further highlighted the superior performance of the CAR method in terms of convergence acceleration, improved predicted correlations, and reduced computational costs. The advancement of the CAR method makes it a highly effective approach, enhancing the applicability of FEP in drug discovery.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


