Saher Gul Ahdi, Javeria Raza Alvi, Azeem Ashfaq, Tipu Sultan
{"title":"小儿神经细胞类脂膜炎:揭示临床和遗传规范。","authors":"Saher Gul Ahdi, Javeria Raza Alvi, Azeem Ashfaq, Tipu Sultan","doi":"10.12669/pjms.40.8.8006","DOIUrl":null,"url":null,"abstract":"<p><strong>Objective: </strong>To unravel the clinical and genetic specifications of Neuronal ceroid lipofuscinosis (NCL).</p><p><strong>Methods: </strong>This is a retrospective cross-sectional study conducted in the Department of Pediatric Neurology Children Hospital and University of Child Health Sciences, Lahore, Pakistan from March 2017 to March 2022. The primary outcome was to measure genotype-phenotype correlation by segregation of phenotypes according to genotype. The secondary outcomes included a correlation between genotype and distribution of age(s) of onset.</p><p><strong>Results: </strong>One hundred fifty three patients clinically diagnosed with NCL underwent genetic testing and pathologic mutation was identified in 32.7% of patients. About 59.6% were male and 37.2% had an affected sibling. The median age was 5.46±1.95 years at the onset of the first symptom i.e., myoclonic seizures in 68%, and motor difficulty in 24%. Other features found were global developmental delay (56%), hypotonia (23%), visual impairment (80%), ataxia (22%), and disc pallor (56%). The most common type was CLN6 (Ceroid lipofuscinosis neuronal) (42%), CLN2 (16%) followed by CLN7 (12%). When 50 patients with recognized mutations were compared with 103 patients with no mutation, family history (p=0.049), early visual loss (p=0.016), hypotonia (p=0.001), white matter signals (p=0.026) and pan-atrophy(p=0.047) was statistically significant in the genetically confirmed NCL. Multiple pairwise comparisons indicated that the estimated age of onset for the CLN1 and CLN2 mutation group was significantly lower than other genotypes including CLN6 (p 0.012), CLN10 (p 0.007) and CLN12 (p 0.007).</p><p><strong>Conclusion: </strong>Following a detailed review of NCL symptomatology, a clinically-oriented approach should be used for a rapid diagnosis with confirmation by targeted molecular testing for future genetic counseling.</p>","PeriodicalId":19958,"journal":{"name":"Pakistan Journal of Medical Sciences","volume":"40 8","pages":"1638-1643"},"PeriodicalIF":1.2000,"publicationDate":"2024-09-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11395386/pdf/","citationCount":"0","resultStr":"{\"title\":\"Pediatric onset neuronal ceroid lipofuscinoses: Unraveling clinical and genetic specifications.\",\"authors\":\"Saher Gul Ahdi, Javeria Raza Alvi, Azeem Ashfaq, Tipu Sultan\",\"doi\":\"10.12669/pjms.40.8.8006\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Objective: </strong>To unravel the clinical and genetic specifications of Neuronal ceroid lipofuscinosis (NCL).</p><p><strong>Methods: </strong>This is a retrospective cross-sectional study conducted in the Department of Pediatric Neurology Children Hospital and University of Child Health Sciences, Lahore, Pakistan from March 2017 to March 2022. The primary outcome was to measure genotype-phenotype correlation by segregation of phenotypes according to genotype. The secondary outcomes included a correlation between genotype and distribution of age(s) of onset.</p><p><strong>Results: </strong>One hundred fifty three patients clinically diagnosed with NCL underwent genetic testing and pathologic mutation was identified in 32.7% of patients. About 59.6% were male and 37.2% had an affected sibling. The median age was 5.46±1.95 years at the onset of the first symptom i.e., myoclonic seizures in 68%, and motor difficulty in 24%. Other features found were global developmental delay (56%), hypotonia (23%), visual impairment (80%), ataxia (22%), and disc pallor (56%). The most common type was CLN6 (Ceroid lipofuscinosis neuronal) (42%), CLN2 (16%) followed by CLN7 (12%). When 50 patients with recognized mutations were compared with 103 patients with no mutation, family history (p=0.049), early visual loss (p=0.016), hypotonia (p=0.001), white matter signals (p=0.026) and pan-atrophy(p=0.047) was statistically significant in the genetically confirmed NCL. Multiple pairwise comparisons indicated that the estimated age of onset for the CLN1 and CLN2 mutation group was significantly lower than other genotypes including CLN6 (p 0.012), CLN10 (p 0.007) and CLN12 (p 0.007).</p><p><strong>Conclusion: </strong>Following a detailed review of NCL symptomatology, a clinically-oriented approach should be used for a rapid diagnosis with confirmation by targeted molecular testing for future genetic counseling.</p>\",\"PeriodicalId\":19958,\"journal\":{\"name\":\"Pakistan Journal of Medical Sciences\",\"volume\":\"40 8\",\"pages\":\"1638-1643\"},\"PeriodicalIF\":1.2000,\"publicationDate\":\"2024-09-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11395386/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Pakistan Journal of Medical Sciences\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.12669/pjms.40.8.8006\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"MEDICINE, GENERAL & INTERNAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Pakistan Journal of Medical Sciences","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.12669/pjms.40.8.8006","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"MEDICINE, GENERAL & INTERNAL","Score":null,"Total":0}
Pediatric onset neuronal ceroid lipofuscinoses: Unraveling clinical and genetic specifications.
Objective: To unravel the clinical and genetic specifications of Neuronal ceroid lipofuscinosis (NCL).
Methods: This is a retrospective cross-sectional study conducted in the Department of Pediatric Neurology Children Hospital and University of Child Health Sciences, Lahore, Pakistan from March 2017 to March 2022. The primary outcome was to measure genotype-phenotype correlation by segregation of phenotypes according to genotype. The secondary outcomes included a correlation between genotype and distribution of age(s) of onset.
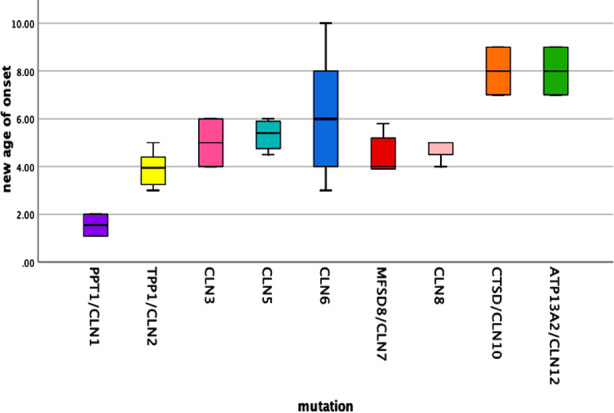
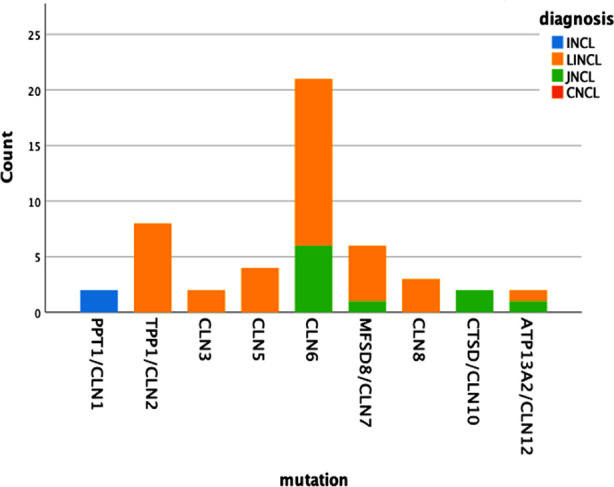
Results: One hundred fifty three patients clinically diagnosed with NCL underwent genetic testing and pathologic mutation was identified in 32.7% of patients. About 59.6% were male and 37.2% had an affected sibling. The median age was 5.46±1.95 years at the onset of the first symptom i.e., myoclonic seizures in 68%, and motor difficulty in 24%. Other features found were global developmental delay (56%), hypotonia (23%), visual impairment (80%), ataxia (22%), and disc pallor (56%). The most common type was CLN6 (Ceroid lipofuscinosis neuronal) (42%), CLN2 (16%) followed by CLN7 (12%). When 50 patients with recognized mutations were compared with 103 patients with no mutation, family history (p=0.049), early visual loss (p=0.016), hypotonia (p=0.001), white matter signals (p=0.026) and pan-atrophy(p=0.047) was statistically significant in the genetically confirmed NCL. Multiple pairwise comparisons indicated that the estimated age of onset for the CLN1 and CLN2 mutation group was significantly lower than other genotypes including CLN6 (p 0.012), CLN10 (p 0.007) and CLN12 (p 0.007).
Conclusion: Following a detailed review of NCL symptomatology, a clinically-oriented approach should be used for a rapid diagnosis with confirmation by targeted molecular testing for future genetic counseling.
期刊介绍:
It is a peer reviewed medical journal published regularly since 1984. It was previously known as quarterly "SPECIALIST" till December 31st 1999. It publishes original research articles, review articles, current practices, short communications & case reports. It attracts manuscripts not only from within Pakistan but also from over fifty countries from abroad.
Copies of PJMS are sent to all the import medical libraries all over Pakistan and overseas particularly in South East Asia and Asia Pacific besides WHO EMRO Region countries. Eminent members of the medical profession at home and abroad regularly contribute their write-ups, manuscripts in our publications. We pursue an independent editorial policy, which allows an opportunity to the healthcare professionals to express their views without any fear or favour. That is why many opinion makers among the medical and pharmaceutical profession use this publication to communicate their viewpoint.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:



