Idoia Bilbao, Miriam Recalde, Fabrice Daian, José Maria Herranz, María Elizalde, Mercedes Iñarrairaegui, Matteo Canale, Maite G Fernández-Barrena, Andrea Casadei-Gardini, Bruno Sangro, Matías A Ávila, Manuel F Landecho Acha, Carmen Berasain, María Arechederra
{"title":"对肝细胞癌的 CpG 甲基化进行全面的硅学分析,确定了与基因表达无关的组织和肿瘤类型特异性标记。","authors":"Idoia Bilbao, Miriam Recalde, Fabrice Daian, José Maria Herranz, María Elizalde, Mercedes Iñarrairaegui, Matteo Canale, Maite G Fernández-Barrena, Andrea Casadei-Gardini, Bruno Sangro, Matías A Ávila, Manuel F Landecho Acha, Carmen Berasain, María Arechederra","doi":"10.1007/s13105-024-01045-8","DOIUrl":null,"url":null,"abstract":"<p><p>DNA methylation is crucial for chromatin structure, transcription regulation and genome stability, defining cellular identity. Aberrant hypermethylation of CpG-rich regions is common in cancer, influencing gene expression. However, the specific contributions of individual epigenetic modifications to tumorigenesis remain under investigation. In hepatocellular carcinoma (HCC), DNA methylation alterations are documented as in other tumor types. We aimed to identify hypermethylated CpGs in HCC, assess their specificity across other tumor types, and investigate their impact on gene expression. To this end, public methylomes from HCC, other liver diseases, and 27 tumor types as well as expression data from TCGA-LIHC and GTEx were analyzed. This study identified 39 CpG sites that were hypermethylated in HCC compared to control liver tissue, and were located within promoter, gene bodies, and intergenic CpG islands. Notably, these CpGs were predominantly unmethylated in healthy liver tissue and other normal tissues. Comparative analysis with 27 other tumors revealed both common and HCC-specific hypermethylated CpGs. Interestingly, the HCC-hypermethylated genes showed minimal expression in the different healthy tissues, with marginal changes in the level of expression in the corresponding tumors. These findings confirm previous evidence on the limited influence of DNA hypermethylation on gene expression regulation in cancer. It also highlights the existence of mechanisms that allow the selection of tissue-specific methylation marks in normally unexpressed genes during carcinogenesis. Overall, our study contributes to demonstrate the complexity of cancer epigenetics, emphasizing the need of better understanding the interplay between DNA methylation, gene expression dynamics, and tumorigenesis.</p>","PeriodicalId":16779,"journal":{"name":"Journal of physiology and biochemistry","volume":" ","pages":"865-879"},"PeriodicalIF":4.3000,"publicationDate":"2024-11-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11682006/pdf/","citationCount":"0","resultStr":"{\"title\":\"Comprehensive in silico CpG methylation analysis in hepatocellular carcinoma identifies tissue- and tumor-type specific marks disconnected from gene expression.\",\"authors\":\"Idoia Bilbao, Miriam Recalde, Fabrice Daian, José Maria Herranz, María Elizalde, Mercedes Iñarrairaegui, Matteo Canale, Maite G Fernández-Barrena, Andrea Casadei-Gardini, Bruno Sangro, Matías A Ávila, Manuel F Landecho Acha, Carmen Berasain, María Arechederra\",\"doi\":\"10.1007/s13105-024-01045-8\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>DNA methylation is crucial for chromatin structure, transcription regulation and genome stability, defining cellular identity. Aberrant hypermethylation of CpG-rich regions is common in cancer, influencing gene expression. However, the specific contributions of individual epigenetic modifications to tumorigenesis remain under investigation. In hepatocellular carcinoma (HCC), DNA methylation alterations are documented as in other tumor types. We aimed to identify hypermethylated CpGs in HCC, assess their specificity across other tumor types, and investigate their impact on gene expression. To this end, public methylomes from HCC, other liver diseases, and 27 tumor types as well as expression data from TCGA-LIHC and GTEx were analyzed. This study identified 39 CpG sites that were hypermethylated in HCC compared to control liver tissue, and were located within promoter, gene bodies, and intergenic CpG islands. Notably, these CpGs were predominantly unmethylated in healthy liver tissue and other normal tissues. Comparative analysis with 27 other tumors revealed both common and HCC-specific hypermethylated CpGs. Interestingly, the HCC-hypermethylated genes showed minimal expression in the different healthy tissues, with marginal changes in the level of expression in the corresponding tumors. These findings confirm previous evidence on the limited influence of DNA hypermethylation on gene expression regulation in cancer. It also highlights the existence of mechanisms that allow the selection of tissue-specific methylation marks in normally unexpressed genes during carcinogenesis. Overall, our study contributes to demonstrate the complexity of cancer epigenetics, emphasizing the need of better understanding the interplay between DNA methylation, gene expression dynamics, and tumorigenesis.</p>\",\"PeriodicalId\":16779,\"journal\":{\"name\":\"Journal of physiology and biochemistry\",\"volume\":\" \",\"pages\":\"865-879\"},\"PeriodicalIF\":4.3000,\"publicationDate\":\"2024-11-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11682006/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of physiology and biochemistry\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1007/s13105-024-01045-8\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/9/21 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of physiology and biochemistry","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1007/s13105-024-01045-8","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/9/21 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
Comprehensive in silico CpG methylation analysis in hepatocellular carcinoma identifies tissue- and tumor-type specific marks disconnected from gene expression.
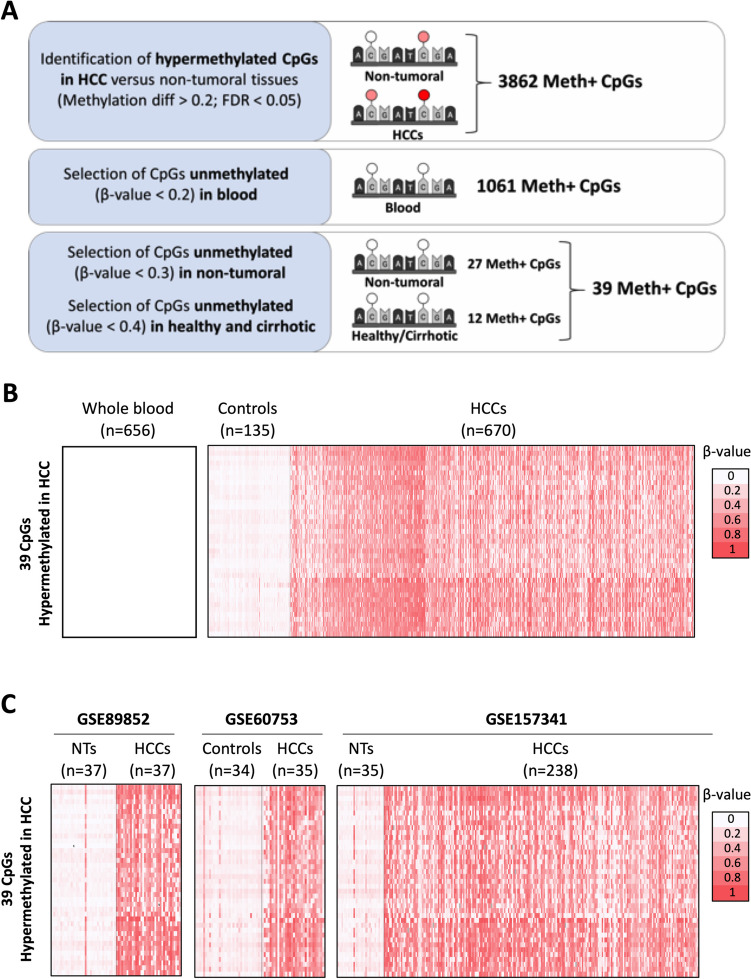
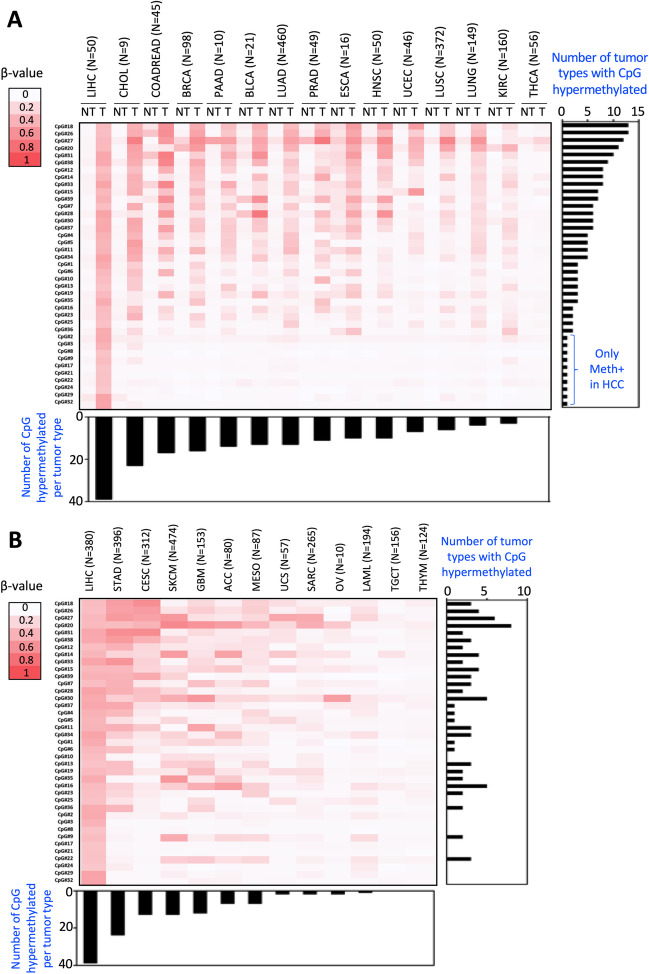
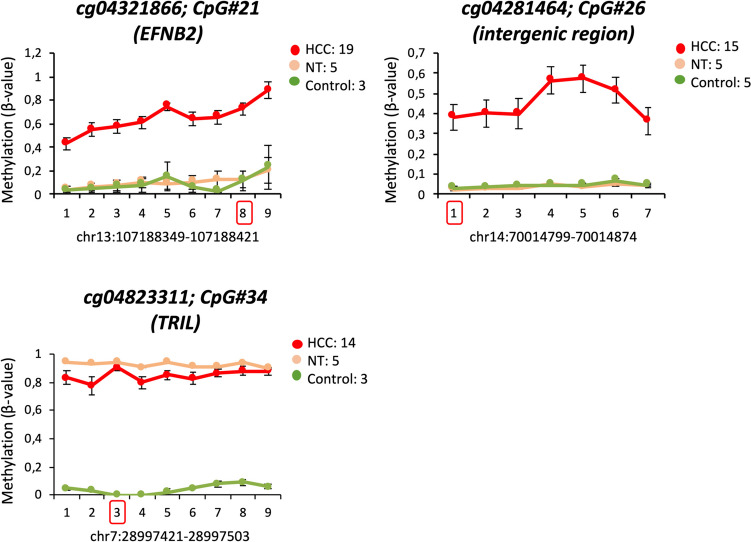
DNA methylation is crucial for chromatin structure, transcription regulation and genome stability, defining cellular identity. Aberrant hypermethylation of CpG-rich regions is common in cancer, influencing gene expression. However, the specific contributions of individual epigenetic modifications to tumorigenesis remain under investigation. In hepatocellular carcinoma (HCC), DNA methylation alterations are documented as in other tumor types. We aimed to identify hypermethylated CpGs in HCC, assess their specificity across other tumor types, and investigate their impact on gene expression. To this end, public methylomes from HCC, other liver diseases, and 27 tumor types as well as expression data from TCGA-LIHC and GTEx were analyzed. This study identified 39 CpG sites that were hypermethylated in HCC compared to control liver tissue, and were located within promoter, gene bodies, and intergenic CpG islands. Notably, these CpGs were predominantly unmethylated in healthy liver tissue and other normal tissues. Comparative analysis with 27 other tumors revealed both common and HCC-specific hypermethylated CpGs. Interestingly, the HCC-hypermethylated genes showed minimal expression in the different healthy tissues, with marginal changes in the level of expression in the corresponding tumors. These findings confirm previous evidence on the limited influence of DNA hypermethylation on gene expression regulation in cancer. It also highlights the existence of mechanisms that allow the selection of tissue-specific methylation marks in normally unexpressed genes during carcinogenesis. Overall, our study contributes to demonstrate the complexity of cancer epigenetics, emphasizing the need of better understanding the interplay between DNA methylation, gene expression dynamics, and tumorigenesis.
期刊介绍:
The Journal of Physiology and Biochemistry publishes original research articles and reviews describing relevant new observations on molecular, biochemical and cellular mechanisms involved in human physiology. All areas of the physiology are covered. Special emphasis is placed on the integration of those levels in the whole-organism. The Journal of Physiology and Biochemistry also welcomes articles on molecular nutrition and metabolism studies, and works related to the genomic or proteomic bases of the physiological functions. Descriptive manuscripts about physiological/biochemical processes or clinical manuscripts will not be considered. The journal will not accept manuscripts testing effects of animal or plant extracts.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


