Zeel Vishnubhai Patel, Priyadarshi Prajjwal, Lakshmi Deepak Bethineedi, Divyakshi J Patel, Kaarvi Khullar, Hinal Patel, Kanishka Khatri, Mohammed Dheyaa Marsool Marsool, Srikanth Gadam, Soumya Aleti, Omniat Amir
{"title":"镰状细胞病治疗的新模式和最新进展:系统回顾。","authors":"Zeel Vishnubhai Patel, Priyadarshi Prajjwal, Lakshmi Deepak Bethineedi, Divyakshi J Patel, Kaarvi Khullar, Hinal Patel, Kanishka Khatri, Mohammed Dheyaa Marsool Marsool, Srikanth Gadam, Soumya Aleti, Omniat Amir","doi":"10.2147/JBM.S477507","DOIUrl":null,"url":null,"abstract":"<p><p>Sickle cell disease (SCD), the most common autosomal recessive genetic disorder, affects the hemoglobin (Hb) chains in human red blood cells. It is caused by mutations in the β-globin genes, leading to the production of hemoglobin S, which results in the formation of sickle-shaped red blood cells (RBCs). These abnormal cells cause hemolysis, endothelial damage, and small vessel occlusion, leading to both acute and long-term complications. According to the World Health Organization's 2008 estimates, SCD affects approximately 2.28 per 1000 individuals globally. Despite this high prevalence, therapeutic advancements have been slow. For many years, the only FDA-approved medications for managing SCD complications were hydroxyurea and deferiprone. However, recent years have seen the approval of several new therapies, including L-glutamine (2017), voxelotor and crizanlizumab (2019), as well as exagamglogene autotemcel (Casgevy) and lovotibeglogene autotemcel (Lyfgenia) (2023). These treatments have proven effective in managing both the acute and chronic effects of SCD, including hemolytic anemia, chronic pain, stroke, vaso-occlusive crises, and multiple organ damage syndromes. This review explores the mechanisms of action, practical considerations, and side effects of these emerging therapies, drawing from a comprehensive search of databases such as PubMed, Medline, and Cochrane.</p>","PeriodicalId":15166,"journal":{"name":"Journal of Blood Medicine","volume":"15 ","pages":"435-447"},"PeriodicalIF":2.7000,"publicationDate":"2024-09-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11404495/pdf/","citationCount":"0","resultStr":"{\"title\":\"Newer Modalities and Updates in the Management of Sickle Cell Disease: A Systematic Review.\",\"authors\":\"Zeel Vishnubhai Patel, Priyadarshi Prajjwal, Lakshmi Deepak Bethineedi, Divyakshi J Patel, Kaarvi Khullar, Hinal Patel, Kanishka Khatri, Mohammed Dheyaa Marsool Marsool, Srikanth Gadam, Soumya Aleti, Omniat Amir\",\"doi\":\"10.2147/JBM.S477507\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Sickle cell disease (SCD), the most common autosomal recessive genetic disorder, affects the hemoglobin (Hb) chains in human red blood cells. It is caused by mutations in the β-globin genes, leading to the production of hemoglobin S, which results in the formation of sickle-shaped red blood cells (RBCs). These abnormal cells cause hemolysis, endothelial damage, and small vessel occlusion, leading to both acute and long-term complications. According to the World Health Organization's 2008 estimates, SCD affects approximately 2.28 per 1000 individuals globally. Despite this high prevalence, therapeutic advancements have been slow. For many years, the only FDA-approved medications for managing SCD complications were hydroxyurea and deferiprone. However, recent years have seen the approval of several new therapies, including L-glutamine (2017), voxelotor and crizanlizumab (2019), as well as exagamglogene autotemcel (Casgevy) and lovotibeglogene autotemcel (Lyfgenia) (2023). These treatments have proven effective in managing both the acute and chronic effects of SCD, including hemolytic anemia, chronic pain, stroke, vaso-occlusive crises, and multiple organ damage syndromes. This review explores the mechanisms of action, practical considerations, and side effects of these emerging therapies, drawing from a comprehensive search of databases such as PubMed, Medline, and Cochrane.</p>\",\"PeriodicalId\":15166,\"journal\":{\"name\":\"Journal of Blood Medicine\",\"volume\":\"15 \",\"pages\":\"435-447\"},\"PeriodicalIF\":2.7000,\"publicationDate\":\"2024-09-12\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11404495/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Blood Medicine\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.2147/JBM.S477507\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q3\",\"JCRName\":\"HEMATOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Blood Medicine","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.2147/JBM.S477507","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/1/1 0:00:00","PubModel":"eCollection","JCR":"Q3","JCRName":"HEMATOLOGY","Score":null,"Total":0}
Newer Modalities and Updates in the Management of Sickle Cell Disease: A Systematic Review.
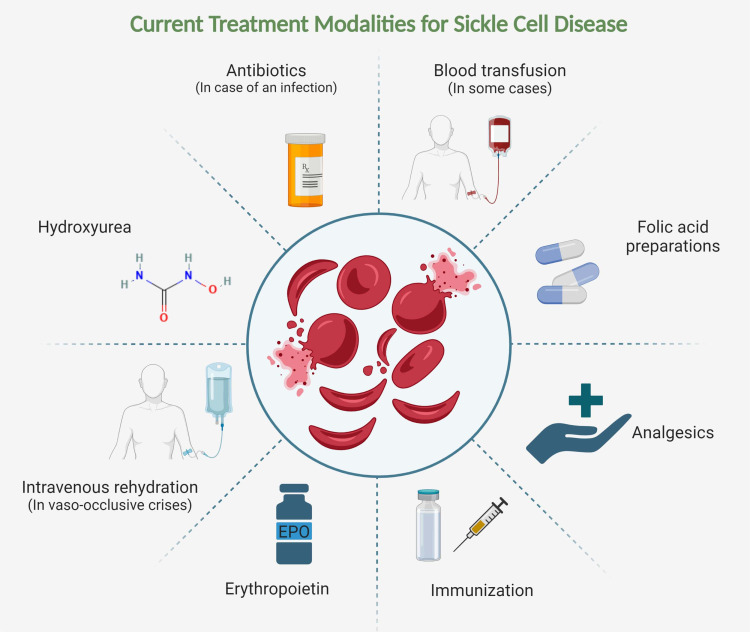
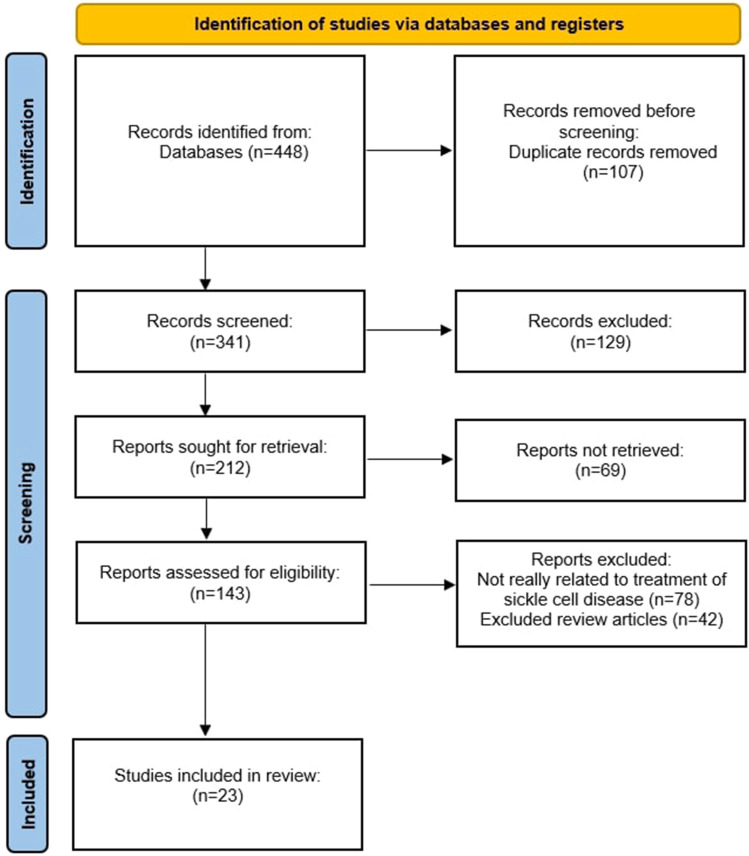
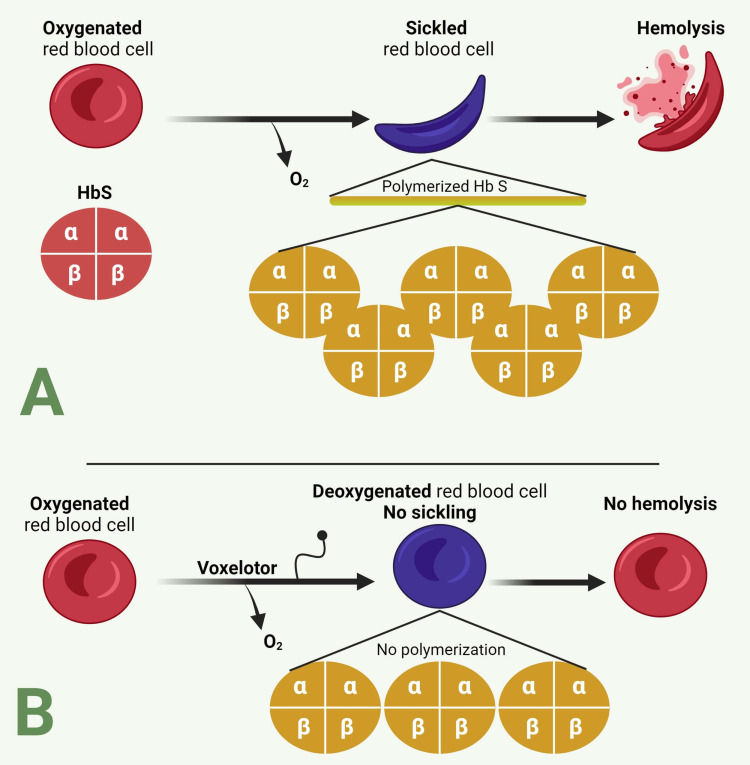
Sickle cell disease (SCD), the most common autosomal recessive genetic disorder, affects the hemoglobin (Hb) chains in human red blood cells. It is caused by mutations in the β-globin genes, leading to the production of hemoglobin S, which results in the formation of sickle-shaped red blood cells (RBCs). These abnormal cells cause hemolysis, endothelial damage, and small vessel occlusion, leading to both acute and long-term complications. According to the World Health Organization's 2008 estimates, SCD affects approximately 2.28 per 1000 individuals globally. Despite this high prevalence, therapeutic advancements have been slow. For many years, the only FDA-approved medications for managing SCD complications were hydroxyurea and deferiprone. However, recent years have seen the approval of several new therapies, including L-glutamine (2017), voxelotor and crizanlizumab (2019), as well as exagamglogene autotemcel (Casgevy) and lovotibeglogene autotemcel (Lyfgenia) (2023). These treatments have proven effective in managing both the acute and chronic effects of SCD, including hemolytic anemia, chronic pain, stroke, vaso-occlusive crises, and multiple organ damage syndromes. This review explores the mechanisms of action, practical considerations, and side effects of these emerging therapies, drawing from a comprehensive search of databases such as PubMed, Medline, and Cochrane.
期刊介绍:
The Journal of Blood Medicine is an international, peer-reviewed, open access, online journal publishing laboratory, experimental and clinical aspects of all topics pertaining to blood based medicine including but not limited to: Transfusion Medicine (blood components, stem cell transplantation, apheresis, gene based therapeutics), Blood collection, Donor issues, Transmittable diseases, and Blood banking logistics, Immunohematology, Artificial and alternative blood based therapeutics, Hematology including disorders/pathology related to leukocytes/immunology, red cells, platelets and hemostasis, Biotechnology/nanotechnology of blood related medicine, Legal aspects of blood medicine, Historical perspectives. Original research, short reports, reviews, case reports and commentaries are invited.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


