Jessika B. Iwanski, Christopher T. Pappas, Rachel M. Mayfield, Gerrie P. Farman, Rebecca Ahrens-Nicklas, Jared M. Churko, Carol C. Gregorio
{"title":"Leiomodin 2 基因突变导致新生儿扩张型心肌病肌动蛋白基因特征改变和心肌细胞功能障碍","authors":"Jessika B. Iwanski, Christopher T. Pappas, Rachel M. Mayfield, Gerrie P. Farman, Rebecca Ahrens-Nicklas, Jared M. Churko, Carol C. Gregorio","doi":"10.1038/s41536-024-00366-y","DOIUrl":null,"url":null,"abstract":"<p>Neonatal dilated cardiomyopathy (DCM) is a poorly understood muscular disease of the heart. Several homozygous biallelic variants in <i>LMOD2</i>, the gene encoding the actin-binding protein Leiomodin 2, have been identified to result in severe DCM. Collectively, <i>LMOD2</i>-related cardiomyopathies present with cardiac dilation and decreased heart contractility, often resulting in neonatal death. Thus, it is evident that Lmod2 is essential to normal human cardiac muscle function. This study aimed to understand the underlying pathophysiology and signaling pathways related to the first reported <i>LMOD2</i> variant (c.1193 G > A, p.Trp398*). Using patient-specific human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) and a mouse model harboring the homologous mutation to the patient, we discovered dysregulated actin-thin filament lengths, altered contractility and calcium handling properties, as well as alterations in the serum response factor (SRF)-dependent signaling pathway. These findings reveal that LMOD2 may be regulating SRF activity in an actin-dependent manner and provide a potential new strategy for the development of biologically active molecules to target <i>LMOD2</i>-related cardiomyopathies.</p>","PeriodicalId":54236,"journal":{"name":"npj Regenerative Medicine","volume":"10 1","pages":""},"PeriodicalIF":6.5000,"publicationDate":"2024-09-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Leiomodin 2 neonatal dilated cardiomyopathy mutation results in altered actin gene signatures and cardiomyocyte dysfunction\",\"authors\":\"Jessika B. Iwanski, Christopher T. Pappas, Rachel M. Mayfield, Gerrie P. Farman, Rebecca Ahrens-Nicklas, Jared M. Churko, Carol C. Gregorio\",\"doi\":\"10.1038/s41536-024-00366-y\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Neonatal dilated cardiomyopathy (DCM) is a poorly understood muscular disease of the heart. Several homozygous biallelic variants in <i>LMOD2</i>, the gene encoding the actin-binding protein Leiomodin 2, have been identified to result in severe DCM. Collectively, <i>LMOD2</i>-related cardiomyopathies present with cardiac dilation and decreased heart contractility, often resulting in neonatal death. Thus, it is evident that Lmod2 is essential to normal human cardiac muscle function. This study aimed to understand the underlying pathophysiology and signaling pathways related to the first reported <i>LMOD2</i> variant (c.1193 G > A, p.Trp398*). Using patient-specific human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) and a mouse model harboring the homologous mutation to the patient, we discovered dysregulated actin-thin filament lengths, altered contractility and calcium handling properties, as well as alterations in the serum response factor (SRF)-dependent signaling pathway. These findings reveal that LMOD2 may be regulating SRF activity in an actin-dependent manner and provide a potential new strategy for the development of biologically active molecules to target <i>LMOD2</i>-related cardiomyopathies.</p>\",\"PeriodicalId\":54236,\"journal\":{\"name\":\"npj Regenerative Medicine\",\"volume\":\"10 1\",\"pages\":\"\"},\"PeriodicalIF\":6.5000,\"publicationDate\":\"2024-09-16\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"npj Regenerative Medicine\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1038/s41536-024-00366-y\",\"RegionNum\":1,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CELL & TISSUE ENGINEERING\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"npj Regenerative Medicine","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1038/s41536-024-00366-y","RegionNum":1,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CELL & TISSUE ENGINEERING","Score":null,"Total":0}
Leiomodin 2 neonatal dilated cardiomyopathy mutation results in altered actin gene signatures and cardiomyocyte dysfunction
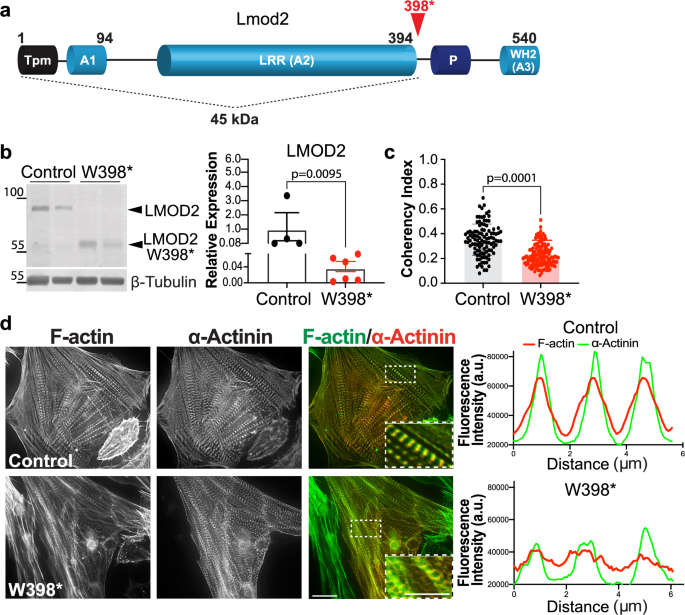
Neonatal dilated cardiomyopathy (DCM) is a poorly understood muscular disease of the heart. Several homozygous biallelic variants in LMOD2, the gene encoding the actin-binding protein Leiomodin 2, have been identified to result in severe DCM. Collectively, LMOD2-related cardiomyopathies present with cardiac dilation and decreased heart contractility, often resulting in neonatal death. Thus, it is evident that Lmod2 is essential to normal human cardiac muscle function. This study aimed to understand the underlying pathophysiology and signaling pathways related to the first reported LMOD2 variant (c.1193 G > A, p.Trp398*). Using patient-specific human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) and a mouse model harboring the homologous mutation to the patient, we discovered dysregulated actin-thin filament lengths, altered contractility and calcium handling properties, as well as alterations in the serum response factor (SRF)-dependent signaling pathway. These findings reveal that LMOD2 may be regulating SRF activity in an actin-dependent manner and provide a potential new strategy for the development of biologically active molecules to target LMOD2-related cardiomyopathies.
期刊介绍:
Regenerative Medicine, an innovative online-only journal, aims to advance research in the field of repairing and regenerating damaged tissues and organs within the human body. As a part of the prestigious Nature Partner Journals series and in partnership with ARMI, this high-quality, open access journal serves as a platform for scientists to explore effective therapies that harness the body's natural regenerative capabilities. With a focus on understanding the fundamental mechanisms of tissue damage and regeneration, npj Regenerative Medicine actively encourages studies that bridge the gap between basic research and clinical tissue repair strategies.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


