利用深度图网络准确预测溶液中自由基反应的壁垒高度
IF 2.8
2区 化学
Q3 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
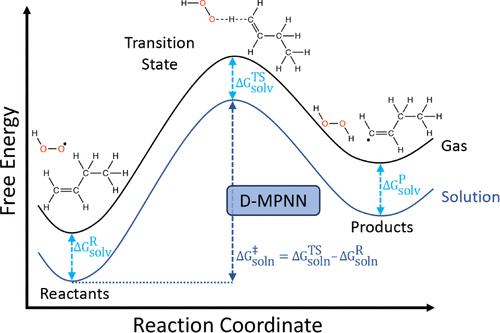
对反应壁垒和溶剂效应的定量估计对于建立动力学机制和预测反应结果至关重要。在此,我们创建了一个新的数据集,其中包含使用 M06-2X/def2-QZVP//B3LYP-D3(BJ)/def2-TZVP 理论水平计算的 5,600 个独特的基本自由基反应。使用 TPSS/def2-TZVP 对每个物种进行了构象搜索。使用 COSMO-RS 计算溶解效应,获得了这些自由基反应在 40 种常见溶剂中的活化和反应吉布斯自由能。这些平衡反应涉及 H、C、N、O 和 S 元素,包含多达 19 个重原子,并具有原子映射 SMILES。所有过渡态都通过内在反应坐标计算进行了验证。接下来,我们训练一个深度图网络,仅使用反应物和生成物的原子映射 SMILES 以及溶剂的 SMILES,直接估算气相和溶液相的活化和反应吉布斯自由能。这种简单的输入表示法避免了在推理过程中对反应物、过渡态和产物结构进行昂贵的计算优化,使我们的模型非常适合高通量预测化学,并能快速为(逆)合成规划工具提供信息。为了正确衡量模型的性能,我们报告了内推和外推数据拆分的结果,并与几个基线模型进行了比较。在训练和测试过程中,我们增加了数据集,包括每个反应的反向和具有不同共振结构的变体。数据扩充后,我们有大约 200 万个条目来训练模型,该模型在溶液中活化吉布斯自由能的测试集平均绝对误差为 1.16 kcal mol-1。我们预计该模型将加快高通量筛选的预测速度,从而快速识别溶液中的相关反应,我们的数据集将作为未来研究的基准。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Accurately Predicting Barrier Heights for Radical Reactions in Solution Using Deep Graph Networks
Quantitative estimates of reaction barriers and solvent effects are essential for developing kinetic mechanisms and predicting reaction outcomes. Here, we create a new data set of 5,600 unique elementary radical reactions calculated using the M06-2X/def2-QZVP//B3LYP-D3(BJ)/def2-TZVP level of theory. A conformer search is done for each species using TPSS/def2-TZVP. Gibbs free energies of activation and of reaction for these radical reactions in 40 common solvents are obtained using COSMO-RS for solvation effects. These balanced reactions involve the elements H, C, N, O, and S, contain up to 19 heavy atoms, and have atom-mapped SMILES. All transition states are verified by an intrinsic reaction coordinate calculation. We next train a deep graph network to directly estimate the Gibbs free energy of activation and of reaction in both gas and solution phases using only the atom-mapped SMILES of the reactant and product and the SMILES of the solvent. This simple input representation avoids computationally expensive optimizations for the reactant, transition state, and product structures during inference, making our model well-suited for high-throughput predictive chemistry and quickly providing information for (retro-)synthesis planning tools. To properly measure model performance, we report results on both interpolative and extrapolative data splits and also compare to several baseline models. During training and testing, the data set is augmented by including the reverse direction of each reaction and variants with different resonance structures. After data augmentation, we have around 2 million entries to train the model, which achieves a testing set mean absolute error of 1.16 kcal mol–1 for the Gibbs free energy of activation in solution. We anticipate this model will accelerate predictions for high-throughput screening to quickly identify relevant reactions in solution, and our data set will serve as a benchmark for future studies.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

The Journal of Physical Chemistry A
化学-物理:原子、分子和化学物理
CiteScore
5.20
自引率
10.30%
发文量
922
审稿时长
1.3 months
期刊介绍:
The Journal of Physical Chemistry A is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


