尿素力场的高斯过程回归建模
IF 2.7
2区 化学
Q3 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
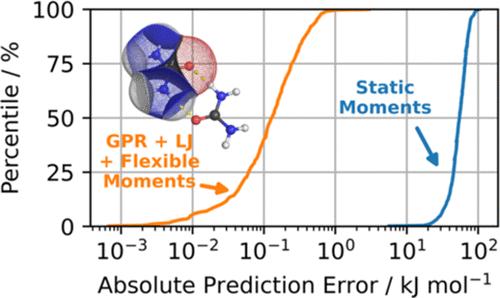
FFLUX 是建立在量子化学拓扑学、高斯过程回归和(高阶)多极静电学三大基石之上的新一代机器学习力场。与标准的自证分子动力学相比,它能以更低的计算成本进行近量子精度的分子动力学研究。FFLUX 以前的研究涉及水和甲酰胺。在这项研究中,我们更进一步,挑战 FFLUX,为尿素这个更大、更灵活的系统建模。结果,我们在 B3LYP/aug-cc-pVTZ 理论水平上训练了尿素模型,其平均绝对误差为 0.4 kJ mol-1,最大预测误差低于 7.0 kJ mol-1。为了测试它们在分子动力学模拟中的性能,我们进行了两组 FFLUX 几何优化:5 个对应能量最小值的二聚体和 75 个随机二聚体。这 5 个二聚体与它们的 ab initio 参照物的均方根偏差低于 0.1 Å。在 75 个随机二聚体中,68% 的二聚体与在 ab initio 水平上得到的二聚体在性质上相同。此外,我们还将 5 个经过 FFLUX 优化的二聚体按照其相对 FFLUX 单点能量的顺序进行了排序,并与 ab initio 方法进行了比较。除了两个连续最小值之间的一次交叉之外,能量排序完全一致。最后,我们证明了与几何相关的(即灵活的)多极矩的重要性,表明缺乏多极矩灵活性会导致分子间总静电能的平均误差超过 2 个数量级。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Toward Gaussian Process Regression Modeling of a Urea Force Field
FFLUX is a next-generation, machine-learnt force field built on three cornerstones: quantum chemical topology, Gaussian process regression, and (high-rank) multipolar electrostatics. It is capable of performing molecular dynamics with near-quantum accuracy at a lower computational cost than standard ab initio molecular dynamics. Previous work with FFLUX was concerned with water and formamide. In this study, we go one step further and challenge FFLUX to model urea, a larger and more flexible system. In result, we have trained urea models at the B3LYP/aug-cc-pVTZ level of theory, with a mean absolute error of 0.4 kJ mol–1 and a maximum prediction error below 7.0 kJ mol–1. To test their performance in molecular dynamics simulations, two sets of FFLUX geometry optimizations were carried out: 5 dimers corresponding to energy minima and 75 random dimers. The 5 dimers were recovered with a root-mean-square deviation below 0.1 Å with respect to their ab initio references. Out of the 75 random dimers, 68% converged to the qualitatively same dimer as those obtained at the ab initio level. Furthermore, we have ranked the 5 FFLUX-optimized dimers in the order of their relative FFLUX single-point energies and compared them with the ab initio method. The energy ranking fully agreed but for one crossover between two successive minima. Finally, we have demonstrated the importance of geometry-dependent (i.e., flexible) multipole moments, showing that the lack of multipole moment flexibility can lead to average errors in the total intermolecular electrostatic energy of more than 2 orders of magnitude.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

The Journal of Physical Chemistry A
化学-物理:原子、分子和化学物理
CiteScore
5.20
自引率
10.30%
发文量
922
审稿时长
1.3 months
期刊介绍:
The Journal of Physical Chemistry A is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


