Caroline Harmon, Austin Bui, Jasmin M. Espejo, Marc Gancayco, Jennifer M. Le, Juan Rangel, Daryl K. Eggers
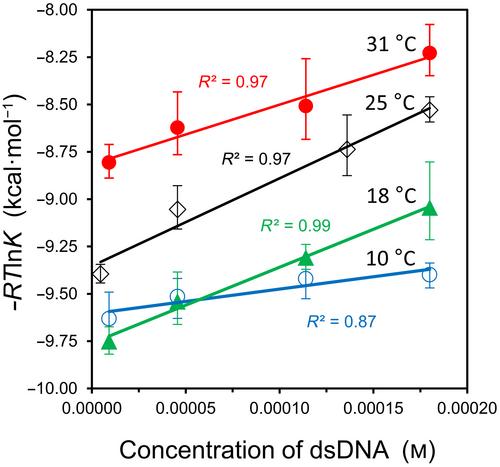
{"title":"DNA 杂交、蛋白质-配体结合和蛋白质折叠的调控方程中的溶解自由能","authors":"Caroline Harmon, Austin Bui, Jasmin M. Espejo, Marc Gancayco, Jennifer M. Le, Juan Rangel, Daryl K. Eggers","doi":"10.1002/2211-5463.13897","DOIUrl":null,"url":null,"abstract":"<p>This work examines the thermodynamics of model biomolecular interactions using a governing equation that accounts for the participation of bulk water in the equilibria. In the first example, the binding affinities of two DNA duplexes, one of nine and one of 10 base pairs in length, are measured and characterized by isothermal titration calorimetry (ITC) as a function of concentration. The results indicate that the change in solvation free energy that accompanies duplex formation (Δ<i>G</i><sup>S</sup>) is large and unfavorable. The duplex with the larger number of G:C pairings yields the largest change in solvation free energy, Δ<i>G</i><sup>S</sup> = +460 kcal·mol<sup>−1</sup>per base pair at 25 °C. A van't Hoff analysis of the data is complicated by the varying degree of intramolecular base stacking within each DNA strand as a function of temperature. A modeling study reveals how the solvation free energy alters the output of a typical ITC experiment and leads to a good, though misleading, fit to the classical equilibrium equation. The same thermodynamic framework is applied to a model protein–ligand interaction, the binding of ribonuclease A with the nucleotide inhibitor 3′-UMP, and to a conformational equilibrium, the change in tertiary structure of α-lactalbumin in molar guanidinium chloride solutions. The ribonuclease study yields a value of Δ<i>G</i><sup>S</sup> = +160 kcal·mol<sup>−1</sup>, whereas the folding equilibrium yields Δ<i>G</i><sup>S</sup> ≈ 0, an apparent characteristic of hydrophobic interactions. These examples provide insight on the role of solvation energy in binding equilibria and suggest a pivot in the fundamental application of thermodynamics to solution chemistry.</p>","PeriodicalId":12187,"journal":{"name":"FEBS Open Bio","volume":"14 11","pages":"1837-1850"},"PeriodicalIF":2.8000,"publicationDate":"2024-09-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/2211-5463.13897","citationCount":"0","resultStr":"{\"title\":\"Solvation free energy in governing equations for DNA hybridization, protein–ligand binding, and protein folding\",\"authors\":\"Caroline Harmon, Austin Bui, Jasmin M. Espejo, Marc Gancayco, Jennifer M. Le, Juan Rangel, Daryl K. Eggers\",\"doi\":\"10.1002/2211-5463.13897\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>This work examines the thermodynamics of model biomolecular interactions using a governing equation that accounts for the participation of bulk water in the equilibria. In the first example, the binding affinities of two DNA duplexes, one of nine and one of 10 base pairs in length, are measured and characterized by isothermal titration calorimetry (ITC) as a function of concentration. The results indicate that the change in solvation free energy that accompanies duplex formation (Δ<i>G</i><sup>S</sup>) is large and unfavorable. The duplex with the larger number of G:C pairings yields the largest change in solvation free energy, Δ<i>G</i><sup>S</sup> = +460 kcal·mol<sup>−1</sup>per base pair at 25 °C. A van't Hoff analysis of the data is complicated by the varying degree of intramolecular base stacking within each DNA strand as a function of temperature. A modeling study reveals how the solvation free energy alters the output of a typical ITC experiment and leads to a good, though misleading, fit to the classical equilibrium equation. The same thermodynamic framework is applied to a model protein–ligand interaction, the binding of ribonuclease A with the nucleotide inhibitor 3′-UMP, and to a conformational equilibrium, the change in tertiary structure of α-lactalbumin in molar guanidinium chloride solutions. The ribonuclease study yields a value of Δ<i>G</i><sup>S</sup> = +160 kcal·mol<sup>−1</sup>, whereas the folding equilibrium yields Δ<i>G</i><sup>S</sup> ≈ 0, an apparent characteristic of hydrophobic interactions. These examples provide insight on the role of solvation energy in binding equilibria and suggest a pivot in the fundamental application of thermodynamics to solution chemistry.</p>\",\"PeriodicalId\":12187,\"journal\":{\"name\":\"FEBS Open Bio\",\"volume\":\"14 11\",\"pages\":\"1837-1850\"},\"PeriodicalIF\":2.8000,\"publicationDate\":\"2024-09-17\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/2211-5463.13897\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"FEBS Open Bio\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/2211-5463.13897\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"FEBS Open Bio","FirstCategoryId":"99","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/2211-5463.13897","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
摘要
这项研究利用一个考虑到散装水参与平衡的控制方程,对模型生物分子相互作用的热力学进行了研究。在第一个例子中,通过等温滴定量热法(ITC)测量并表征了两个 DNA 双链(一个长度为 9 个碱基对,一个长度为 10 个碱基对)的结合亲和力与浓度的函数关系。结果表明,伴随双链体形成的溶解自由能变化(ΔGS)是巨大而不利的。G:C 配对数目较多的双链产生的溶解自由能变化最大,25 °C时每个碱基对的溶解自由能为ΔGS = +460 kcal-mol-1。由于每条 DNA 链的分子内碱基堆叠程度随温度的变化而变化,因此对数据进行 van't Hoff 分析变得非常复杂。建模研究揭示了溶解自由能如何改变典型 ITC 实验的输出结果,并导致与经典平衡方程的良好拟合(尽管会产生误导)。同样的热力学框架还被应用于蛋白质-配体相互作用模型--核糖核酸酶 A 与核苷酸抑制剂 3′-UMP 的结合,以及构象平衡--α-乳白蛋白在摩尔氯化胍溶液中三级结构的变化。核糖核酸酶研究得出的值ΔGS = +160 kcal-mol-1,而折叠平衡得出的值ΔGS ≈ 0,这是疏水相互作用的明显特征。这些例子深入揭示了溶解能在结合平衡中的作用,并为热力学在溶液化学中的基本应用提供了一个支点。
Solvation free energy in governing equations for DNA hybridization, protein–ligand binding, and protein folding
This work examines the thermodynamics of model biomolecular interactions using a governing equation that accounts for the participation of bulk water in the equilibria. In the first example, the binding affinities of two DNA duplexes, one of nine and one of 10 base pairs in length, are measured and characterized by isothermal titration calorimetry (ITC) as a function of concentration. The results indicate that the change in solvation free energy that accompanies duplex formation (ΔGS) is large and unfavorable. The duplex with the larger number of G:C pairings yields the largest change in solvation free energy, ΔGS = +460 kcal·mol−1per base pair at 25 °C. A van't Hoff analysis of the data is complicated by the varying degree of intramolecular base stacking within each DNA strand as a function of temperature. A modeling study reveals how the solvation free energy alters the output of a typical ITC experiment and leads to a good, though misleading, fit to the classical equilibrium equation. The same thermodynamic framework is applied to a model protein–ligand interaction, the binding of ribonuclease A with the nucleotide inhibitor 3′-UMP, and to a conformational equilibrium, the change in tertiary structure of α-lactalbumin in molar guanidinium chloride solutions. The ribonuclease study yields a value of ΔGS = +160 kcal·mol−1, whereas the folding equilibrium yields ΔGS ≈ 0, an apparent characteristic of hydrophobic interactions. These examples provide insight on the role of solvation energy in binding equilibria and suggest a pivot in the fundamental application of thermodynamics to solution chemistry.
期刊介绍:
FEBS Open Bio is an online-only open access journal for the rapid publication of research articles in molecular and cellular life sciences in both health and disease. The journal''s peer review process focuses on the technical soundness of papers, leaving the assessment of their impact and importance to the scientific community.
FEBS Open Bio is owned by the Federation of European Biochemical Societies (FEBS), a not-for-profit organization, and is published on behalf of FEBS by FEBS Press and Wiley. Any income from the journal will be used to support scientists through fellowships, courses, travel grants, prizes and other FEBS initiatives.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


