通过深度学习揭示人类病原体中的蛋白质相互作用
IF 20.5
1区 生物学
Q1 MICROBIOLOGY
引用次数: 0
摘要
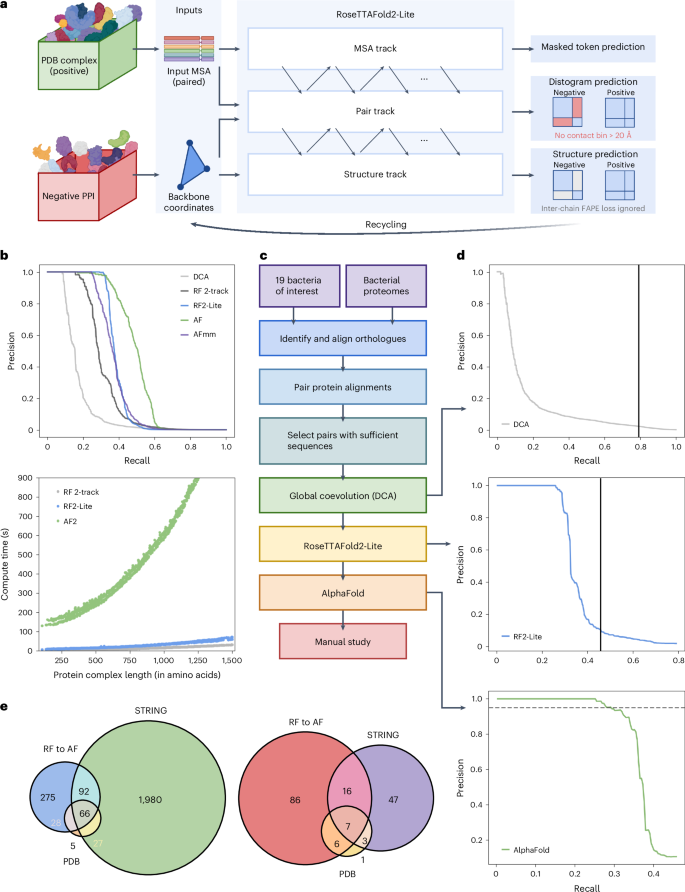
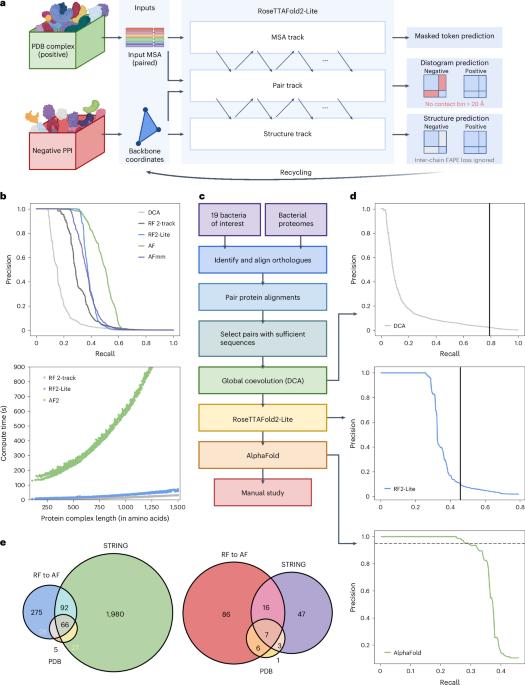
鉴定细菌蛋白质-蛋白质相互作用并预测这些复合物的结构有助于了解致病机制和开发传染性疾病的治疗方法。在这里,我们开发了 RoseTTAFold2-Lite,它是一种快速深度学习模型,利用残基-残基协同进化和蛋白质结构预测,在整个蛋白质组范围内系统地鉴定蛋白质-蛋白质相互作用并从结构上描述其特征。利用这一管道,我们搜索了 19 种人类细菌病原体中的 7800 万对蛋白质,确定了 1923 个涉及重要基因的可信预测复合物和 256 个涉及毒力因子的复合物。其中许多复合体以前并不为人所知;我们对 12 个这样的预测进行了实验测试,其中一半得到了验证。预测的相互作用跨越了核心代谢和毒力途径,从转录后修饰到酸中和再到外膜机制,它们将有助于我们了解这些重要病原体的生物学特性,并设计出对抗它们的药物。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Protein interactions in human pathogens revealed through deep learning
Identification of bacterial protein–protein interactions and predicting the structures of these complexes could aid in the understanding of pathogenicity mechanisms and developing treatments for infectious diseases. Here we developed RoseTTAFold2-Lite, a rapid deep learning model that leverages residue–residue coevolution and protein structure prediction to systematically identify and structurally characterize protein–protein interactions at the proteome-wide scale. Using this pipeline, we searched through 78 million pairs of proteins across 19 human bacterial pathogens and identified 1,923 confidently predicted complexes involving essential genes and 256 involving virulence factors. Many of these complexes were not previously known; we experimentally tested 12 such predictions, and half of them were validated. The predicted interactions span core metabolic and virulence pathways ranging from post-transcriptional modification to acid neutralization to outer-membrane machinery and should contribute to our understanding of the biology of these important pathogens and the design of drugs to combat them. RoseTTAFold2-Lite uses residue–residue coevolution and protein structure prediction to identify and structurally characterize protein–protein interactions in bacterial pathogens.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Nature Microbiology
Immunology and Microbiology-Microbiology
CiteScore
44.40
自引率
1.10%
发文量
226
期刊介绍:
Nature Microbiology aims to cover a comprehensive range of topics related to microorganisms. This includes:
Evolution: The journal is interested in exploring the evolutionary aspects of microorganisms. This may include research on their genetic diversity, adaptation, and speciation over time.
Physiology and cell biology: Nature Microbiology seeks to understand the functions and characteristics of microorganisms at the cellular and physiological levels. This may involve studying their metabolism, growth patterns, and cellular processes.
Interactions: The journal focuses on the interactions microorganisms have with each other, as well as their interactions with hosts or the environment. This encompasses investigations into microbial communities, symbiotic relationships, and microbial responses to different environments.
Societal significance: Nature Microbiology recognizes the societal impact of microorganisms and welcomes studies that explore their practical applications. This may include research on microbial diseases, biotechnology, or environmental remediation.
In summary, Nature Microbiology is interested in research related to the evolution, physiology and cell biology of microorganisms, their interactions, and their societal relevance.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


