完全多极模型作为多体相互作用的一般框架,以水为例证
IF 5.7
1区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
我们介绍了多体力场的一般框架--完全多极模型(CMM),它利用电子密度指数衰减调制的多极电矩作为分子间相互作用能量分解分析所有项的通用函数形式。有了这种共同的函数形式,CMM 模型建立了表述清晰的阻尼张量,可在长程和短程达到正确的渐近线,同时从形式上确保不会出现短程灾难。CMM 描述了具有短程各向异性的分散、交换极化和保利斥力等可分离的 EDA 项,极化为分子内电荷波动和诱导偶极子,而电荷转移则描述了分子间明确的电荷移动,并通过耦合到极化方程自然地描述了多体电荷转移。我们还利用一种新的单体电势,通过对莫尔斯电势进行电场校正来考虑分子内极化,以确保 CMM 重现所有与物理相关的单体特性,包括偶极矩、分子极化率以及偶极和极化率导数。CMM 的质量通过 EDA 单项的一致性和对大量水簇数据验证集的能量和几何形状的出色推断得到了证明。本文章由计算机程序翻译,如有差异,请以英文原文为准。
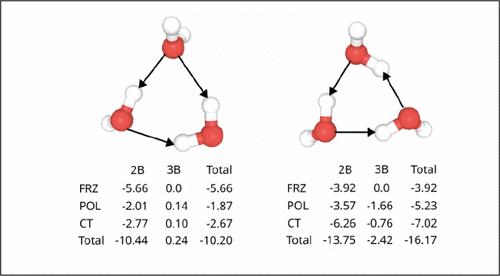
Completely Multipolar Model as a General Framework for Many-Body Interactions as Illustrated for Water
We introduce a general framework for many-body force fields, the Completely Multipolar Model (CMM), that utilizes multipolar electrical moments modulated by exponential decay of electron density as a common functional form for all terms of an energy decomposition analysis of intermolecular interactions. With this common functional form, the CMM model establishes well-formulated damped tensors that reach the correct asymptotes at both long- and short-range while formally ensuring no short-range catastrophes. CMM describes the separable EDA terms of dispersion, exchange polarization, and Pauli repulsion with short-ranged anisotropy, polarization as intramolecular charge fluctuations and induced dipoles, while charge transfer describes explicit movement of charge between molecules, and naturally describes many-body charge transfer by coupling into the polarization equations. We also utilize a new one-body potential that accounts for intramolecular polarization by including an electric field-dependent correction to the Morse potential to ensure that CMM reproduces all physically relevant monomer properties including the dipole moment, molecular polarizability, and dipole and polarizability derivatives. The quality of CMM is illustrated through agreement of individual terms of the EDA and excellent extrapolation to energies and geometries of an extensive validation set of water cluster data.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Journal of Chemical Theory and Computation
化学-物理:原子、分子和化学物理
CiteScore
9.90
自引率
16.40%
发文量
568
审稿时长
1 months
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


