提取生物分子构象转变反应坐标的模式演化指标
IF 5.5
1区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
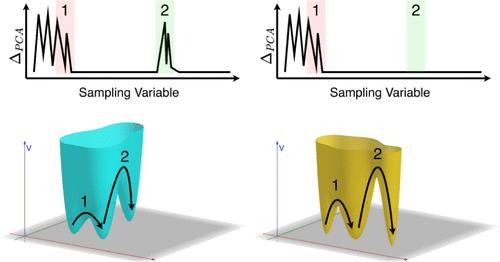
生物大分子具有复杂的多维能谱,因此提取合适的、非直观的集体变量(CV)来描述其构象转变具有挑战性。目前,人们采用降维方法和机器学习(ML)方案,从分子动力学(MD)/蒙特卡罗(MC)轨迹或生物大分子结构数据库中获取集体变量。然而,生成能准确描述底层能谱的可靠 CV 的最低采样条件仍不清楚。在此,我们通过开发一种模式演化指标(MeM)来解决这一问题,以从非平衡 MD/MC 轨迹中提取可精确定位新状态并描述参考最小值附近局部转变的 CV。我们为统计降维和机器学习方法提出了 MeM 的一般数学公式。将 MeM 应用于模型势能图的 MC 轨迹和溶解丙氨酸二肽的 MD 轨迹时发现,在参考最小值附近定位新状态的主成分在相关状态之间的轨迹局部平衡之前就已出现。最后,我们展示了 MeM 在设计高效的偏置采样方案中的可能应用,以构建连接各状态之间转换的精确能量景观切片。MeM 有助于加快在生物分子构象状态周围寻找新的最小值,并能准确估计能量景观上的状态的热力学以及描述相关的转变。本文章由计算机程序翻译,如有差异,请以英文原文为准。
A Mode Evolution Metric to Extract Reaction Coordinates for Biomolecular Conformational Transitions
The complex, multidimensional energy landscape of biomolecules makes the extraction of suitable, nonintuitive collective variables (CVs) that describe their conformational transitions challenging. At present, dimensionality reduction approaches and machine learning (ML) schemes are employed to obtain CVs from molecular dynamics (MD)/Monte Carlo (MC) trajectories or structural databanks for biomolecules. However, minimum sampling conditions to generate reliable CVs that accurately describe the underlying energy landscape remain unclear. Here, we address this issue by developing a Mode evolution Metric (MeM) to extract CVs that can pinpoint new states and describe local transitions in the vicinity of a reference minimum from nonequilibrated MD/MC trajectories. We present a general mathematical formulation of MeM for both statistical dimensionality reduction and machine learning approaches. Application of MeM to MC trajectories of model potential energy landscapes and MD trajectories of solvated alanine dipeptide reveals that the principal components which locate new states in the vicinity of a reference minimum emerge well before the trajectories locally equilibrate between the associated states. Finally, we demonstrate a possible application of MeM in designing efficient biased sampling schemes to construct accurate energy landscape slices that link transitions between states. MeM can help speed up the search for new minima around a biomolecular conformational state and enable the accurate estimation of thermodynamics for states lying on the energy landscape and the description of associated transitions.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Journal of Chemical Theory and Computation
化学-物理:原子、分子和化学物理
CiteScore
9.90
自引率
16.40%
发文量
568
审稿时长
1 months
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


