利用 MolSpin 中的随机薛定谔方程研究自由基对的自旋动力学
IF 5.5
1区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
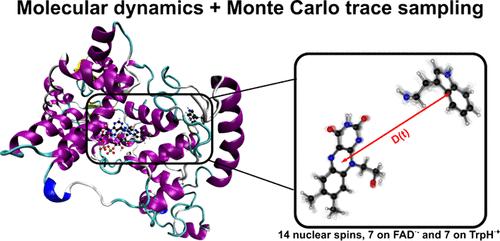
自由基对的化学反应性受到电子自旋和核自旋相互作用的强烈影响。要详细了解这些影响,需要对自旋动力学进行量子描述,其中要考虑自旋相关反应速率、与外部磁场的相互作用、自旋-自旋相互作用以及与波动环境耦合造成的自旋相干性损失。真实的化学和生化系统经常涉及具有多核自旋系统的自由基,这给建模带来了严峻的计算挑战。在这里,我们在软件包 MolSpin 中实现了一种基于随机薛定谔方程的方法。大型电子-核自旋系统可以通过不对称自旋选择性重组反应、各向异性超精细相互作用以及非零交换和偶极耦合进行高效模拟。利用分子动力学和量子化学计算所确定的自旋相互作用的随机时间依赖性,或通过允许速率系数明确依赖于时间,可以对自旋松弛进行建模。这种方法的灵活性为探索复杂分子运动对化学转化自旋动力学的影响开辟了新途径。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Spin Dynamics of Radical Pairs Using the Stochastic Schrödinger Equation in MolSpin
The chemical reactivity of radical pairs is strongly influenced by the interactions of electronic and nuclear spins. A detailed understanding of these effects requires a quantum description of the spin dynamics that considers spin-dependent reaction rates, interactions with external magnetic fields, spin–spin interactions, and the loss of spin coherence caused by coupling to a fluctuating environment. Modeling real chemical and biochemical systems, which frequently involve radicals with multinuclear spin systems, poses a severe computational challenge. Here, we implement a method based on the stochastic Schrödinger equation in the software package MolSpin. Large electron–nuclear spin systems can be simulated efficiently, with asymmetric spin-selective recombination reactions, anisotropic hyperfine interactions, and nonzero exchange and dipolar couplings. Spin-relaxation can be modeled using the stochastic time-dependence of spin interactions determined by molecular dynamics and quantum chemical calculations or by allowing rate coefficients to be explicitly time-dependent. The flexibility afforded by this approach opens new avenues for exploring the effects of complex molecular motions on the spin dynamics of chemical transformations.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Journal of Chemical Theory and Computation
化学-物理:原子、分子和化学物理
CiteScore
9.90
自引率
16.40%
发文量
568
审稿时长
1 months
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


