{"title":"利用第一原理计算富锑 InBixSb1-x 合金的晶格常数、带隙能、吸收系数和介电函数","authors":"Chuan-Zhen Zhao, Yu-Ting Dai","doi":"10.1016/j.jpcs.2024.112307","DOIUrl":null,"url":null,"abstract":"<div><p>So far, little is known about the electronic and optical properties of InBi<sub>x</sub>Sb<sub>1-x</sub>. In this work, the first-principles calculations are performed to research the lattice constant, the bandgap energy, the absorption coefficient and the dielectric function of the antimony-rich InBi<sub>x</sub>Sb<sub>1-x</sub>. The results show that the bowing coefficient for the lattice constant is merely −0.012 Å. According to the band structures, it is found that the Sb-rich InBi<sub>x</sub>Sb<sub>1-x</sub> possesses a direct bandgap at G point. Its bandgap reduction is due to the ascending of the valence band maximum (VBM) and the descending of the conduction band minimum (CBM). For the sake of providing a good description for the bandgap energy in the Sb-rich range, the modified valence band anticrossing (MVBAC) model plus a linear equation is utilized. The result shows that the predicted positive to negative bandgap transition occurs at x = 0.13. Besides, the valence band offset between InSb and InBi is identified to be 0.27eV. For the optical properties, it is found that the Bi component is effective in raising the static dielectric constant of InBi<sub>x</sub>Sb<sub>1-x</sub>. However, it has a minor effect on the transition ability of the electrons. In the Sb-rich component range, the critical point energies <span><math><mrow><msub><mi>E</mi><mn>0</mn></msub><mo>+</mo><msub><mo>Δ</mo><mn>0</mn></msub></mrow></math></span> <span><math><mrow><msub><mi>E</mi><mn>1</mn></msub></mrow></math></span>, <span><math><mrow><msub><mi>E</mi><mn>1</mn></msub><mo>+</mo><msub><mo>Δ</mo><mn>1</mn></msub></mrow></math></span> <span><math><mrow><msub><mi>E</mi><mn>2</mn></msub></mrow></math></span> and <span><math><mrow><msubsup><mi>E</mi><mn>1</mn><mo>′</mo></msubsup></mrow></math></span> are found to shift toward the low energy direction. The result of the absorption spectra supports this opinion as increasing Bi component leads to a redshift of the absorption spectra. The adjustable bandgap energy and the optical properties of InBi<sub>x</sub>Sb<sub>1-x</sub> demonstrate that it is a strong candidate for fabricating the photodetectors in the 8–12 μm spectral region.</p></div>","PeriodicalId":16811,"journal":{"name":"Journal of Physics and Chemistry of Solids","volume":"196 ","pages":"Article 112307"},"PeriodicalIF":4.3000,"publicationDate":"2024-09-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Lattice constant, bandgap energy, absorption coefficient and dielectric function of the antimony-rich InBixSb1-x alloy using first-principles calculations\",\"authors\":\"Chuan-Zhen Zhao, Yu-Ting Dai\",\"doi\":\"10.1016/j.jpcs.2024.112307\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>So far, little is known about the electronic and optical properties of InBi<sub>x</sub>Sb<sub>1-x</sub>. In this work, the first-principles calculations are performed to research the lattice constant, the bandgap energy, the absorption coefficient and the dielectric function of the antimony-rich InBi<sub>x</sub>Sb<sub>1-x</sub>. The results show that the bowing coefficient for the lattice constant is merely −0.012 Å. According to the band structures, it is found that the Sb-rich InBi<sub>x</sub>Sb<sub>1-x</sub> possesses a direct bandgap at G point. Its bandgap reduction is due to the ascending of the valence band maximum (VBM) and the descending of the conduction band minimum (CBM). For the sake of providing a good description for the bandgap energy in the Sb-rich range, the modified valence band anticrossing (MVBAC) model plus a linear equation is utilized. The result shows that the predicted positive to negative bandgap transition occurs at x = 0.13. Besides, the valence band offset between InSb and InBi is identified to be 0.27eV. For the optical properties, it is found that the Bi component is effective in raising the static dielectric constant of InBi<sub>x</sub>Sb<sub>1-x</sub>. However, it has a minor effect on the transition ability of the electrons. In the Sb-rich component range, the critical point energies <span><math><mrow><msub><mi>E</mi><mn>0</mn></msub><mo>+</mo><msub><mo>Δ</mo><mn>0</mn></msub></mrow></math></span> <span><math><mrow><msub><mi>E</mi><mn>1</mn></msub></mrow></math></span>, <span><math><mrow><msub><mi>E</mi><mn>1</mn></msub><mo>+</mo><msub><mo>Δ</mo><mn>1</mn></msub></mrow></math></span> <span><math><mrow><msub><mi>E</mi><mn>2</mn></msub></mrow></math></span> and <span><math><mrow><msubsup><mi>E</mi><mn>1</mn><mo>′</mo></msubsup></mrow></math></span> are found to shift toward the low energy direction. The result of the absorption spectra supports this opinion as increasing Bi component leads to a redshift of the absorption spectra. The adjustable bandgap energy and the optical properties of InBi<sub>x</sub>Sb<sub>1-x</sub> demonstrate that it is a strong candidate for fabricating the photodetectors in the 8–12 μm spectral region.</p></div>\",\"PeriodicalId\":16811,\"journal\":{\"name\":\"Journal of Physics and Chemistry of Solids\",\"volume\":\"196 \",\"pages\":\"Article 112307\"},\"PeriodicalIF\":4.3000,\"publicationDate\":\"2024-09-11\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Physics and Chemistry of Solids\",\"FirstCategoryId\":\"88\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S0022369724004426\",\"RegionNum\":3,\"RegionCategory\":\"材料科学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Physics and Chemistry of Solids","FirstCategoryId":"88","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0022369724004426","RegionNum":3,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
Lattice constant, bandgap energy, absorption coefficient and dielectric function of the antimony-rich InBixSb1-x alloy using first-principles calculations
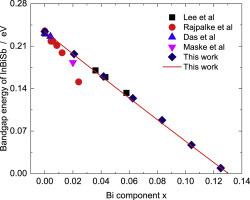
So far, little is known about the electronic and optical properties of InBixSb1-x. In this work, the first-principles calculations are performed to research the lattice constant, the bandgap energy, the absorption coefficient and the dielectric function of the antimony-rich InBixSb1-x. The results show that the bowing coefficient for the lattice constant is merely −0.012 Å. According to the band structures, it is found that the Sb-rich InBixSb1-x possesses a direct bandgap at G point. Its bandgap reduction is due to the ascending of the valence band maximum (VBM) and the descending of the conduction band minimum (CBM). For the sake of providing a good description for the bandgap energy in the Sb-rich range, the modified valence band anticrossing (MVBAC) model plus a linear equation is utilized. The result shows that the predicted positive to negative bandgap transition occurs at x = 0.13. Besides, the valence band offset between InSb and InBi is identified to be 0.27eV. For the optical properties, it is found that the Bi component is effective in raising the static dielectric constant of InBixSb1-x. However, it has a minor effect on the transition ability of the electrons. In the Sb-rich component range, the critical point energies , and are found to shift toward the low energy direction. The result of the absorption spectra supports this opinion as increasing Bi component leads to a redshift of the absorption spectra. The adjustable bandgap energy and the optical properties of InBixSb1-x demonstrate that it is a strong candidate for fabricating the photodetectors in the 8–12 μm spectral region.
期刊介绍:
The Journal of Physics and Chemistry of Solids is a well-established international medium for publication of archival research in condensed matter and materials sciences. Areas of interest broadly include experimental and theoretical research on electronic, magnetic, spectroscopic and structural properties as well as the statistical mechanics and thermodynamics of materials. The focus is on gaining physical and chemical insight into the properties and potential applications of condensed matter systems.
Within the broad scope of the journal, beyond regular contributions, the editors have identified submissions in the following areas of physics and chemistry of solids to be of special current interest to the journal:
Low-dimensional systems
Exotic states of quantum electron matter including topological phases
Energy conversion and storage
Interfaces, nanoparticles and catalysts.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


