Tianhang Zhou , Chen Shen , Zhenghao Wu , Xingying Lan , Yi Xiao
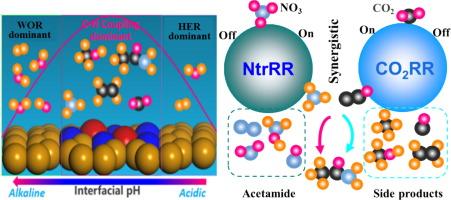
{"title":"通过 pH 依赖性离子和诱导离子调节界面反应环境,了解在双铜基掺杂 N 的碳催化剂上将 NO3- 集成 CO2 还原成碳氮化合物的过程中 C-N 键的耦合性能","authors":"Tianhang Zhou , Chen Shen , Zhenghao Wu , Xingying Lan , Yi Xiao","doi":"10.1016/j.jechem.2024.08.049","DOIUrl":null,"url":null,"abstract":"<div><p>Dual atomic catalysts (DAC), particularly copper (Cu<sub>2</sub>)-based nitrogen (N) doped graphene, show great potential to effectively convert CO<sub>2</sub> and nitrate (NO<sub>3</sub><sup>−</sup>) into important industrial chemicals such as ethylene, glycol, acetamide, and urea through an efficient catalytical process that involves C–C and C–N coupling. However, the origin of the coupling activity remained unclear, which substantially hinders the rational design of Cu-based catalysts for the N-integrated CO<sub>2</sub> reduction reaction (CO<sub>2</sub>RR). To address this challenge, this work performed advanced density functional theory calculations incorporating explicit solvation based on a Cu<sub>2</sub>-based N-doped carbon (Cu<sub>2</sub>N<sub>6</sub>C<sub>10</sub>) catalyst for CO<sub>2</sub>RR. These calculations are aimed to gain insight into the reaction mechanisms for the synthesis of ethylene, acetamide, and urea via coupling in the interfacial reaction micro-environment. Due to the sluggishness of CO<sub>2</sub>, the formation of a solvation electric layer by anions (F<sup>−</sup>, Cl<sup>−</sup>, Br<sup>−</sup>, and I<sup>−</sup>) and cations (Na<sup>+</sup>, Mg<sup>2+</sup>, K<sup>+</sup>, and Ca<sup>2+</sup>) leads to electron transfer towards the Cu surface. This process significantly accelerates the reduction of CO<sub>2</sub>. These results reveal that *CO intermediates play a pivotal role in N-integrated CO<sub>2</sub>RR. Remarkably, the Cu<sub>2</sub>-based N-doped carbon catalyst examined in this study has demonstrated the most potential for C–N coupling to date. Our findings reveal that through the process of a condensation reaction between *CO and NH<sub>2</sub>OH for urea synthesis, *NO<sub>3</sub><sup>−</sup> is reduced to *NH<sub>3</sub>, and *CO<sub>2</sub> to *CCO at dual Cu atom sites. This dual-site reduction facilitates the synthesis of acetamide through a nucleophilic reaction between NH<sub>3</sub> and the ketene intermediate. Furthermore, we found that the I<sup>−</sup> and Mg<sup>2+</sup> ions, influenced by pH, were highly effective for acetamide and ammonia synthesis, except when F<sup>−</sup> and Ca<sup>2+</sup> were present. Furthermore, the mechanisms of C–N bond formation were investigated via ab-initio molecular dynamics simulations, and we found that adjusting the micro-environment can change the dominant side reaction, shifting from hydrogen production in acidic conditions to water reduction in alkaline ones. This study introduces a novel approach using ion-H<sub>2</sub>O cages to significantly enhance the efficiency of C–N coupling reactions.</p></div>","PeriodicalId":15728,"journal":{"name":"Journal of Energy Chemistry","volume":"100 ","pages":"Pages 273-285"},"PeriodicalIF":13.1000,"publicationDate":"2024-09-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Tuning the interfacial reaction environment via pH-dependent and induced ions to understand C–N bonds coupling performance in NO3− integrated CO2 reduction to carbon and nitrogen compounds over dual Cu-based N-doped carbon catalyst\",\"authors\":\"Tianhang Zhou , Chen Shen , Zhenghao Wu , Xingying Lan , Yi Xiao\",\"doi\":\"10.1016/j.jechem.2024.08.049\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>Dual atomic catalysts (DAC), particularly copper (Cu<sub>2</sub>)-based nitrogen (N) doped graphene, show great potential to effectively convert CO<sub>2</sub> and nitrate (NO<sub>3</sub><sup>−</sup>) into important industrial chemicals such as ethylene, glycol, acetamide, and urea through an efficient catalytical process that involves C–C and C–N coupling. However, the origin of the coupling activity remained unclear, which substantially hinders the rational design of Cu-based catalysts for the N-integrated CO<sub>2</sub> reduction reaction (CO<sub>2</sub>RR). To address this challenge, this work performed advanced density functional theory calculations incorporating explicit solvation based on a Cu<sub>2</sub>-based N-doped carbon (Cu<sub>2</sub>N<sub>6</sub>C<sub>10</sub>) catalyst for CO<sub>2</sub>RR. These calculations are aimed to gain insight into the reaction mechanisms for the synthesis of ethylene, acetamide, and urea via coupling in the interfacial reaction micro-environment. Due to the sluggishness of CO<sub>2</sub>, the formation of a solvation electric layer by anions (F<sup>−</sup>, Cl<sup>−</sup>, Br<sup>−</sup>, and I<sup>−</sup>) and cations (Na<sup>+</sup>, Mg<sup>2+</sup>, K<sup>+</sup>, and Ca<sup>2+</sup>) leads to electron transfer towards the Cu surface. This process significantly accelerates the reduction of CO<sub>2</sub>. These results reveal that *CO intermediates play a pivotal role in N-integrated CO<sub>2</sub>RR. Remarkably, the Cu<sub>2</sub>-based N-doped carbon catalyst examined in this study has demonstrated the most potential for C–N coupling to date. Our findings reveal that through the process of a condensation reaction between *CO and NH<sub>2</sub>OH for urea synthesis, *NO<sub>3</sub><sup>−</sup> is reduced to *NH<sub>3</sub>, and *CO<sub>2</sub> to *CCO at dual Cu atom sites. This dual-site reduction facilitates the synthesis of acetamide through a nucleophilic reaction between NH<sub>3</sub> and the ketene intermediate. Furthermore, we found that the I<sup>−</sup> and Mg<sup>2+</sup> ions, influenced by pH, were highly effective for acetamide and ammonia synthesis, except when F<sup>−</sup> and Ca<sup>2+</sup> were present. Furthermore, the mechanisms of C–N bond formation were investigated via ab-initio molecular dynamics simulations, and we found that adjusting the micro-environment can change the dominant side reaction, shifting from hydrogen production in acidic conditions to water reduction in alkaline ones. This study introduces a novel approach using ion-H<sub>2</sub>O cages to significantly enhance the efficiency of C–N coupling reactions.</p></div>\",\"PeriodicalId\":15728,\"journal\":{\"name\":\"Journal of Energy Chemistry\",\"volume\":\"100 \",\"pages\":\"Pages 273-285\"},\"PeriodicalIF\":13.1000,\"publicationDate\":\"2024-09-04\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Energy Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S2095495624006053\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"Energy\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Energy Chemistry","FirstCategoryId":"92","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2095495624006053","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"Energy","Score":null,"Total":0}
引用次数: 0
摘要
双原子催化剂(DAC),特别是基于铜(Cu2)的氮(N)掺杂石墨烯,通过涉及 C-C 和 C-N 偶联的高效催化过程,显示出将 CO2 和硝酸盐(NO3-)有效转化为乙烯、乙二醇、乙酰胺和尿素等重要工业化学品的巨大潜力。然而,耦合活性的起源仍不清楚,这严重阻碍了用于 N-整合二氧化碳还原反应(CO2RR)的铜基催化剂的合理设计。为了应对这一挑战,本研究以用于 CO2RR 的 Cu2 基 N 掺杂碳 (Cu2N6C10) 催化剂为基础,进行了包含显式溶解的高级密度泛函理论计算。这些计算旨在深入了解在界面反应微环境中通过耦合合成乙烯、乙酰胺和尿素的反应机理。由于 CO2 的迟滞性,阴离子(F-、Cl-、Br- 和 I-)和阳离子(Na+、Mg2+、K+ 和 Ca2+)形成的溶电层导致电子向铜表面转移。这一过程大大加速了 CO2 的还原。这些结果表明,*CO 中间体在整合 N 的 CO2RR 中起着关键作用。值得注意的是,本研究中考察的基于 Cu2 的掺杂 N 的碳催化剂展现了迄今为止 C-N 耦合的最大潜力。我们的研究结果表明,通过*CO 和 NH2OH 之间的缩合反应(用于尿素合成),*NO3- 被还原为*NH3,*CO2 在双 Cu 原子位点上被还原为*CCO。这种双位点还原有助于通过 NH3 与烯酮中间体之间的亲核反应合成乙酰胺。此外,我们发现 I- 和 Mg2+ 离子受 pH 值的影响,对乙酰胺和氨的合成非常有效,但 F- 和 Ca2+ 离子存在时除外。此外,我们还通过非原位分子动力学模拟研究了 C-N 键的形成机制,发现调整微环境可以改变主要的副反应,从酸性条件下的产氢反应转变为碱性条件下的还原水反应。这项研究介绍了一种利用离子-H2O 笼显著提高 C-N 偶联反应效率的新方法。
Tuning the interfacial reaction environment via pH-dependent and induced ions to understand C–N bonds coupling performance in NO3− integrated CO2 reduction to carbon and nitrogen compounds over dual Cu-based N-doped carbon catalyst
Dual atomic catalysts (DAC), particularly copper (Cu2)-based nitrogen (N) doped graphene, show great potential to effectively convert CO2 and nitrate (NO3−) into important industrial chemicals such as ethylene, glycol, acetamide, and urea through an efficient catalytical process that involves C–C and C–N coupling. However, the origin of the coupling activity remained unclear, which substantially hinders the rational design of Cu-based catalysts for the N-integrated CO2 reduction reaction (CO2RR). To address this challenge, this work performed advanced density functional theory calculations incorporating explicit solvation based on a Cu2-based N-doped carbon (Cu2N6C10) catalyst for CO2RR. These calculations are aimed to gain insight into the reaction mechanisms for the synthesis of ethylene, acetamide, and urea via coupling in the interfacial reaction micro-environment. Due to the sluggishness of CO2, the formation of a solvation electric layer by anions (F−, Cl−, Br−, and I−) and cations (Na+, Mg2+, K+, and Ca2+) leads to electron transfer towards the Cu surface. This process significantly accelerates the reduction of CO2. These results reveal that *CO intermediates play a pivotal role in N-integrated CO2RR. Remarkably, the Cu2-based N-doped carbon catalyst examined in this study has demonstrated the most potential for C–N coupling to date. Our findings reveal that through the process of a condensation reaction between *CO and NH2OH for urea synthesis, *NO3− is reduced to *NH3, and *CO2 to *CCO at dual Cu atom sites. This dual-site reduction facilitates the synthesis of acetamide through a nucleophilic reaction between NH3 and the ketene intermediate. Furthermore, we found that the I− and Mg2+ ions, influenced by pH, were highly effective for acetamide and ammonia synthesis, except when F− and Ca2+ were present. Furthermore, the mechanisms of C–N bond formation were investigated via ab-initio molecular dynamics simulations, and we found that adjusting the micro-environment can change the dominant side reaction, shifting from hydrogen production in acidic conditions to water reduction in alkaline ones. This study introduces a novel approach using ion-H2O cages to significantly enhance the efficiency of C–N coupling reactions.
期刊介绍:
The Journal of Energy Chemistry, the official publication of Science Press and the Dalian Institute of Chemical Physics, Chinese Academy of Sciences, serves as a platform for reporting creative research and innovative applications in energy chemistry. It mainly reports on creative researches and innovative applications of chemical conversions of fossil energy, carbon dioxide, electrochemical energy and hydrogen energy, as well as the conversions of biomass and solar energy related with chemical issues to promote academic exchanges in the field of energy chemistry and to accelerate the exploration, research and development of energy science and technologies.
This journal focuses on original research papers covering various topics within energy chemistry worldwide, including:
Optimized utilization of fossil energy
Hydrogen energy
Conversion and storage of electrochemical energy
Capture, storage, and chemical conversion of carbon dioxide
Materials and nanotechnologies for energy conversion and storage
Chemistry in biomass conversion
Chemistry in the utilization of solar energy

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


