MMFA-DTA:用于药物-靶点亲和力预测的多模态特征注意融合网络,用于抗击 SARS-CoV-2 的药物再利用
IF 5.7
1区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
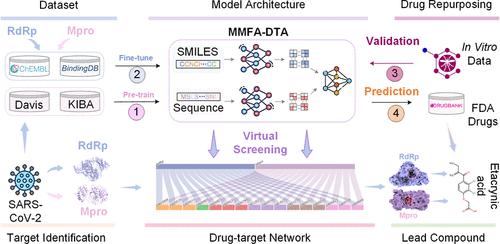
新型传染病的不断出现对全球公共卫生安全构成了重大威胁,因此有必要开发直接针对病原体的小分子抑制剂。SARS-CoV-2的RNA依赖性RNA聚合酶(RdRp)和主要蛋白酶(Mpro)已被验证为治疗COVID-19的潜在关键抗病毒药物靶点。然而,传统的新药研发周期长达 10-15 年,无法满足疫情期间的紧急需求。在此,我们提出了一种用于药物再利用的通用多模态深度学习框架--MMFA-DTA,以实现对已知药物的快速虚拟筛选,并显著提高发现效率。通过从小分子和蛋白质中提取图拓扑和序列特征,我们设计了注意机制来实现跨模态的动态融合。结果表明,在戴维斯和 KIBA 数据集上,MMFA-DTA 在药物-靶点亲和力预测方面的表现优于几种最先进的基线方法,验证了异构信息整合在表征学习和相互作用建模方面的优势。对 COVID-19 相关生物活性数据的进一步微调增强了对关键 SARS-CoV-2 酶的模型预测。案例研究筛选了 FDA 批准的药物库,成功地确定了依他昔酸(etacrynic acid)作为抗 RdRp 和 Mpro 的潜在先导化合物。分子动力学模拟进一步证实了依他昔酸对这些靶点的稳定性和结合亲和力。这项研究证明了深度学习和药物再利用策略在支持抗病毒药物发现方面的巨大潜力和优势。所提出的通用快速响应计算框架对于防范未来的公共卫生事件具有重要意义。本文章由计算机程序翻译,如有差异,请以英文原文为准。
MMFA-DTA: Multimodal Feature Attention Fusion Network for Drug-Target Affinity Prediction for Drug Repurposing Against SARS-CoV-2
The continuous emergence of novel infectious diseases poses a significant threat to global public health security, necessitating the development of small-molecule inhibitors that directly target pathogens. The RNA-dependent RNA polymerase (RdRp) and main protease (Mpro) of SARS-CoV-2 have been validated as potential key antiviral drug targets for the treatment of COVID-19. However, the conventional new drug R&D cycle takes 10–15 years, failing to meet the urgent needs during epidemics. Here, we propose a general multimodal deep learning framework for drug repurposing, MMFA-DTA, to enable rapid virtual screening of known drugs and significantly improve discovery efficiency. By extracting graph topological and sequence features from both small molecules and proteins, we design attention mechanisms to achieve dynamic fusion across modalities. Results demonstrate the superior performance of MMFA-DTA in drug-target affinity prediction over several state-of-the-art baseline methods on Davis and KIBA data sets, validating the benefits of heterogeneous information integration for representation learning and interaction modeling. Further fine-tuning on COVID-19-relevant bioactivity data enhances model predictions for critical SARS-CoV-2 enzymes. Case studies screening the FDA-approved drug library successfully identify etacrynic acid as the potential lead compound against both RdRp and Mpro. Molecular dynamics simulations further confirm the stability and binding affinity of etacrynic acid to these targets. This study proves the great potential and advantages of deep learning and drug repurposing strategies in supporting antiviral drug discovery. The proposed general and rapid response computational framework holds significance for preparedness against future public health events.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Journal of Chemical Theory and Computation
化学-物理:原子、分子和化学物理
CiteScore
9.90
自引率
16.40%
发文量
568
审稿时长
1 months
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


