利用可解释图注意网络预测汽化特性,设计绿色化学品
IF 11.3
1区 化学
Q1 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
对汽化特性的计算预测有助于绿色化学品的全新设计,包括清洁替代燃料、用于高效热能回收的工作液以及易于降解和回收的聚合物。在此,我们开发了化学可解释图注意网络,用于预测与可再生能源利用性能相关的五种物理特性:汽化热(HoV)、临界温度、闪点、沸点和液体热容量。考虑到其不确定性和温度依赖性,HoV 的预测模型使用了 150 000 个数据点进行训练。接下来,通过迁移学习将该模型扩展到其他属性,以克服数据点较少(700-7500 个)所带来的局限性。然后对模型的化学可解释性进行了研究,证明该模型可以解释分子结构对汽化特性的影响。最后,开发的预测模型被应用于设计具有理想特性的化学物质,如高效绿色工作液、燃料和聚合物,从而在实验前实现快速准确的筛选。本文章由计算机程序翻译,如有差异,请以英文原文为准。
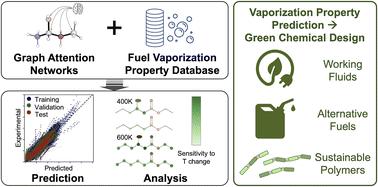
Designing green chemicals by predicting vaporization properties using explainable graph attention networks†
Computational predictions of vaporization properties aid the de novo design of green chemicals, including clean alternative fuels, working fluids for efficient thermal energy recovery, and polymers that are easily degradable and recyclable. Here, we developed chemically explainable graph attention networks to predict five physical properties pertinent to performance in utilizing renewable energy: heat of vaporization (HoV), critical temperature, flash point, boiling point, and liquid heat capacity. The predictive model for HoV was trained using ∼150 000 data points, considering their uncertainties and temperature dependence. Next, this model was expanded to the other properties through transfer learning to overcome the limitations due to fewer data points (700–7500). The chemical interpretability of the model was then investigated, demonstrating that the model explains molecular structural effects on vaporization properties. Finally, the developed predictive models were applied to design chemicals that have desirable properties as efficient and green working fluids, fuels, and polymers, enabling fast and accurate screening before experiments.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

ACS Catalysis
CHEMISTRY, PHYSICAL-
CiteScore
20.80
自引率
6.20%
发文量
1253
审稿时长
1.5 months
期刊介绍:
ACS Catalysis is an esteemed journal that publishes original research in the fields of heterogeneous catalysis, molecular catalysis, and biocatalysis. It offers broad coverage across diverse areas such as life sciences, organometallics and synthesis, photochemistry and electrochemistry, drug discovery and synthesis, materials science, environmental protection, polymer discovery and synthesis, and energy and fuels.
The scope of the journal is to showcase innovative work in various aspects of catalysis. This includes new reactions and novel synthetic approaches utilizing known catalysts, the discovery or modification of new catalysts, elucidation of catalytic mechanisms through cutting-edge investigations, practical enhancements of existing processes, as well as conceptual advances in the field. Contributions to ACS Catalysis can encompass both experimental and theoretical research focused on catalytic molecules, macromolecules, and materials that exhibit catalytic turnover.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


