{"title":"输血依赖型β地中海贫血患者使用鲁帕特罗的实际疗效和安全性以及反应的预测因素","authors":"Daniele Lello Panzieri, Dario Consonni, Natalia Scaramellini, Guido Ausenda, Francesca Granata, Nancy Caponio, Lorena Duca, Simona Leoni, Silvia Elli, Marta Ferraresi, Vittorio Bolis, Cristina Curcio, Milena Agata Irrera, Diletta Maira, Giovanna Graziadei, Elena Cassinerio, Maria Domenica Cappellini, Rayan Bou-Fakhredin, Valentina Brancaleoni, Irene Motta","doi":"10.1002/ajh.27474","DOIUrl":null,"url":null,"abstract":"<p>Luspatercept is the first erythropoiesis-modulating agent approved by the Food and Drug Administration (FDA) and the European Medicines Agency (EMA) for treating anemia in adult transfusion-dependent β-thalassemia (TDT) patients. As observed in clinical trials<span><sup>1</sup></span> and real-life experience,<span><sup>2</sup></span> response to luspatercept in TDT is heterogeneous. It can range from patients who do not respond to those who become transfusion-independent. So far, no predictors of response have been identified. However, the definition of the different profiles and predictors of response is necessary for optimizing treatment allocation, limiting costs, and increasing sustainability. The ELEMENT study is an observational prospective cohort study that enrolled adult TDT patients regularly followed at Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico in Milan (Italy) treated with luspatercept. Luspatercept was administered, according to the indications of the Italian Regulatory Agency, subcutaneously at a starting dose of 1.0 mg/kg every 3 weeks and was increased to 1.25 mg/kg after dose 3 if a clinical response was not achieved.<span><sup>3</sup></span> The drug was discontinued according to the Italian Regulatory Agency indications, namely, if the patient does not achieve transfusion reduction (amount not specified) after three doses at the maximum dosage or in the presence of unacceptable toxicity. Treatment response was assessed by comparing the transfusion burden (TB) during any 12-week treatment period with that in the 24 weeks before treatment. Responders (RSP) were defined as individuals who have a reduction of the TB ≥33% in any 12 weeks of treatment, while those with a TB reduction <33% were considered “non-responders” (NR). Moreover, as in the phase 3 trial, transfusion independence was defined as a transfusion-free period of at least 8 weeks.<span><sup>1</sup></span> In this study, TB was also expressed as the number of units of packed red blood cells (pRBC) per week (unit/week) by dividing the number of units transfused in a period by the number of weeks evaluated.</p><p>Between January 1, 2021, and May 31, 2024, 56 TDT patients received at least one dose of luspatercept after drug authorization. At the start of the study, treatment was offered to TDT patients with high TB, iron overload demonstrated by T2* magnetic resonance imaging (MRI), or other clinically relevant conditions that might benefit from TB reduction. Subsequently, the treatment was made available to all the patients who met the Italian regulatory agency criteria and were willing to receive the drug. The decision to initiate treatment was always discussed between the physician and the patient.</p><p>Seven patients discontinued the drug before completing at least 12 weeks. The reasons for discontinuation are detailed in Table S1 (early drug discontinuation group). At the time of the analysis, one patient had not yet completed 12 weeks of treatment.</p><p>Data from the 48 patients who received luspatercept for at least 12 weeks were included, and their characteristics at enrollment are presented in Figure 1A. Out of 48 patients, two paused their treatment temporarily, one due to a personal decision and the other due to an adverse event (AE). They were later re-challenged with the treatment and achieved a response similar to the first period, resulting in both patients attaining transfusion independence. The median age was 41, and 44% (21/48) were females. In this sample, 38% (18/48) were splenectomized. Sixteen out of 48 had a β0/β0 genotype, and one had HbE/β-thalassemia. Overall, 37/48 (77%) patients had a TB >15 units in the 24 weeks prior to treatment initiation. Among these, 12 patients had a TB ranging from 20 to 24 units. The median treatment period was 48 weeks (12–172), and 39 out of 48 (81%) received treatment for at least 24 weeks. Seventeen out of 48 (35%) were responders (Figure S1), of whom 10/48 (21%) showed a TB reduction ≥33% in weeks 13–24 (the primary endpoint of the phase 3 trial). Eleven out of 17 (65%) patients in the RSP group had a TB reduction of ≥50% for at least one 12-week interval. Four patients became transfusion-independent for at least 12 weeks, of whom three for at least 22 consecutive weeks. TB, expressed as unit/week, did not change in the NR group (0.7 unit/week in the 24 weeks before luspatercept vs. 0.7 unit/week during the treatment, <i>p</i> = 0.95), while it decreased in the RSP group (0.8 unit/week in the 24 weeks before luspatercept vs. 0.5 unit/week during the treatment, <i>p</i> = 0.001). Overall, in our sample, the mean pretreatment pre-transfusion Hb (pt-Hb) was similar in both groups (Figure 1A). The pt-Hb increased between pre- and during-treatment in both NR (9.1 ± 0.5 g/dL pretreatment vs. 9.3 ± 0.4 g/dL under treatment, <i>p</i> = 0.03) and RSP (from 9.2 ± 0.6 to 9.5 ± 0.5 g/dL, <i>p</i> = 0.025) groups.</p><p>Twenty-one out of 48 (44%) discontinued treatment after at least 12 weeks, with 16 being NR. The reasons for discontinuation are detailed in Table S1.<span><sup>4</sup></span></p><p>The drug was generally well tolerated, and no deaths occurred. The frequency of AEs was similar to those reported in the phase 3 study, and a detailed list can be found in Table S2.</p><p>RSP and NR did not differ in TB during the 24 weeks before treatment initiation when evaluating the number of transfused units, even though iron intake was slightly higher in the NR (0.32 ± 0.07 vs. 0.27 ± 0.08 mg/kg/day, <i>p</i> = 0.09). Patients were overall well chelated. When comparing the two groups, RSP showed higher liver iron concentration (LIC), in a range considered borderline and in the lower range of optimal chelation therapy. As shown in Figure S2 after 24 weeks of treatment, we observed a greater increase in reticulocyte count in RSP than in NR (0.2, 0.0–1.1 × 10<sup>12</sup>/L vs. 0.1, 0.0–0.5 × 10<sup>12</sup>/L, <i>p</i> = 0.04).</p><p>Fetal hemoglobin (HbF) at baseline was the only clinically relevant hematological parameter significantly different between RSP and NR. Also, the baseline absolute value of HbF (g/dL) provided the best discrimination between RSP and NR after a receiver operating characteristic (ROC) analysis, with an area under the curve (AUC) of 0.82 (95% confidence interval [CI] 0.68–0.96; Figure 1B). Baseline values of reticulocytes did not increase the AUC. The statistical cut-off point of the ROC curve was identified for a baseline absolute value of HbF of 0.6 g/dL, corresponding to a negative predictive value (NPV) of 92% (95% CI: 74–99, 23 NR out of 25 with HbF <0.6 g/dL).</p><p>An increase in HbF was also observed in both groups; however, the RSP reached higher values than the NR at 24 weeks, 21.1% (4.2–59.1; corresponding to the median absolute HbF of 2.1 g/dL) versus 7.6% (3.2–37.1; corresponding to the median absolute value of HbF of 0.7 g/dL), respectively (<i>p</i> = 0.0001). In the random-intercept linear regression analysis, the best fit was achieved using a cubic model to describe the trend of HbF increase over time. Both RSP and NR exhibited a similar pattern, with an initial increase in levels that reached a plateau around week 16 (Figure 1C).</p><p>Currently, data on luspatercept safety, efficacy, and potential predictors of response primarily come from clinical trials.<span><sup>1, 2, 5</sup></span> Here, we present findings from the largest published real-world cohort of TDT patients treated with luspatercept. Our study explores predictors of response, which are essential for clinicians to assess the risk–benefit profile when considering prescribing this agent. In our study, the response rate was similar to that of the phase 3 trial when evaluating weeks 13–24, namely the primary endpoint of the BELIEVE study. However, the overall response rate over any 12-week interval in our cohort was lower (35% vs. about 70% in the phase 3 trial). This difference could be attributed to the shorter median treatment duration in our study (48 vs. 64 weeks). Reasons for this include adherence to regulatory guidelines for drug prescription and the patients' involvement in treatment decisions in real life. This can affect, for example, the identification of late-responders.<span><sup>6</sup></span> In our sample, most patients completed 24 weeks of treatment, which is a reasonable time for expecting a response.<span><sup>7</sup></span> Other differences emerged when comparing our data to those of the trial. Our patients were more heavily transfused, with 77% having a TB >15 units of pRBC in the 24 weeks before the initiation of luspatercept, compared to 43.5% in the BELIEVE trial. Interestingly, we have identified baseline HbF levels as a predictor of response to treatment. A baseline HbF <0.6 g/dL was associated with a lower likelihood of response to luspatercept (8% of RSP having baseline HbF <0.6 g/dL). Conversely, TDT patients who have a baseline HbF of 0.6 g/dL or higher exhibit a high likelihood of responding to treatment (64% of patients with HbF >0.6 g/dL were RSP). Of note, those with a baseline HbF <0.6 g/dL did not have a higher TB before luspatercept (median TB in the HbF <0.6 g/dL group: 19 units vs. median TB in the HbF >0.6 g/dL group: 17 units; <i>p</i> = 0.11). We do not recommend excluding a priori patients with a baseline value of HbF <0.6 g/dL from treatment. Rather, we suggest carefully considering initiating luspatercept in these patients, especially in the presence of an uncertain risk–benefit ratio (e.g., a risk factor for an AE). Interestingly, regardless of the baseline level, all patients treated with luspatercept experienced an increase in HbF. However, responders showed a higher increase in absolute HbF values compared to NRs.</p><p>Additionally, the presence of severe genotypes did not prevent a response, as one-fourth of RSP had a β0/β0 genotype.</p><p>Some limitations in this study should be acknowledged. First, the sample size is limited given the rarity of the disease and the recent approval of luspatercept in Italy. Second, the observation period in this study was shorter than that of the Phase 3 BELIEVE trial.</p><p>In conclusion, this study confirms that the effectiveness of luspatercept in a real-world cohort of TDT patients is similar to the phase 3 clinical trial and identifies HbF at baseline and its trend in the first few weeks of treatment as a predictive marker of response. Further studies are needed to elucidate the differences in baseline HbF levels between responders and NRs.</p><p>IM, VB, and DLP conceived and designed the study. GA, SE, SL, MF, NS, VB, DM, IM, and DLP collected the data. DC, DLP, IM, VB, NS, and NC analyzed the data. VB, RB-F, CC, NC, LD, FG, and MAI performed the experiments. IM, DLP, VB, and RB-F wrote the paper. All the authors revised the paper.</p><p>This work was partially supported by the Italian Ministry of Health and Fondazione IRCCS Ca' Granda (RC_2022/23), the funding grant of the Società Italiana di Medicina Interna (SIMI), the grant PRIN2022—project number 2022L5YWZT.</p><p>MDC has been or is a current consultant for Novartis, Celgene Corp, Bristol Myers Squibb, Vifor Pharma, and Ionis Pharmaceuticals and has received research funding from Novartis, Celgene Corp, Bristol Myers Squibb, La Jolla Pharmaceutical Company, Roche, Protagonist Therapeutics, and CRISPR Therapeutics. IM is a scientific advisory board member of Bristol Myers Squibb and Sanofi and received honoraria for lectures from Bristol Myers Squibb and Sanofi and research support from Sanofi.</p><p>Informed consent was obtained from all individual participants included in the study.</p>","PeriodicalId":7724,"journal":{"name":"American Journal of Hematology","volume":"99 12","pages":"2395-2398"},"PeriodicalIF":10.1000,"publicationDate":"2024-09-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/ajh.27474","citationCount":"0","resultStr":"{\"title\":\"Real-world efficacy and safety of luspatercept and predictive factors of response in patients with transfusion-dependent β-thalassemia\",\"authors\":\"Daniele Lello Panzieri, Dario Consonni, Natalia Scaramellini, Guido Ausenda, Francesca Granata, Nancy Caponio, Lorena Duca, Simona Leoni, Silvia Elli, Marta Ferraresi, Vittorio Bolis, Cristina Curcio, Milena Agata Irrera, Diletta Maira, Giovanna Graziadei, Elena Cassinerio, Maria Domenica Cappellini, Rayan Bou-Fakhredin, Valentina Brancaleoni, Irene Motta\",\"doi\":\"10.1002/ajh.27474\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Luspatercept is the first erythropoiesis-modulating agent approved by the Food and Drug Administration (FDA) and the European Medicines Agency (EMA) for treating anemia in adult transfusion-dependent β-thalassemia (TDT) patients. As observed in clinical trials<span><sup>1</sup></span> and real-life experience,<span><sup>2</sup></span> response to luspatercept in TDT is heterogeneous. It can range from patients who do not respond to those who become transfusion-independent. So far, no predictors of response have been identified. However, the definition of the different profiles and predictors of response is necessary for optimizing treatment allocation, limiting costs, and increasing sustainability. The ELEMENT study is an observational prospective cohort study that enrolled adult TDT patients regularly followed at Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico in Milan (Italy) treated with luspatercept. Luspatercept was administered, according to the indications of the Italian Regulatory Agency, subcutaneously at a starting dose of 1.0 mg/kg every 3 weeks and was increased to 1.25 mg/kg after dose 3 if a clinical response was not achieved.<span><sup>3</sup></span> The drug was discontinued according to the Italian Regulatory Agency indications, namely, if the patient does not achieve transfusion reduction (amount not specified) after three doses at the maximum dosage or in the presence of unacceptable toxicity. Treatment response was assessed by comparing the transfusion burden (TB) during any 12-week treatment period with that in the 24 weeks before treatment. Responders (RSP) were defined as individuals who have a reduction of the TB ≥33% in any 12 weeks of treatment, while those with a TB reduction <33% were considered “non-responders” (NR). Moreover, as in the phase 3 trial, transfusion independence was defined as a transfusion-free period of at least 8 weeks.<span><sup>1</sup></span> In this study, TB was also expressed as the number of units of packed red blood cells (pRBC) per week (unit/week) by dividing the number of units transfused in a period by the number of weeks evaluated.</p><p>Between January 1, 2021, and May 31, 2024, 56 TDT patients received at least one dose of luspatercept after drug authorization. At the start of the study, treatment was offered to TDT patients with high TB, iron overload demonstrated by T2* magnetic resonance imaging (MRI), or other clinically relevant conditions that might benefit from TB reduction. Subsequently, the treatment was made available to all the patients who met the Italian regulatory agency criteria and were willing to receive the drug. The decision to initiate treatment was always discussed between the physician and the patient.</p><p>Seven patients discontinued the drug before completing at least 12 weeks. The reasons for discontinuation are detailed in Table S1 (early drug discontinuation group). At the time of the analysis, one patient had not yet completed 12 weeks of treatment.</p><p>Data from the 48 patients who received luspatercept for at least 12 weeks were included, and their characteristics at enrollment are presented in Figure 1A. Out of 48 patients, two paused their treatment temporarily, one due to a personal decision and the other due to an adverse event (AE). They were later re-challenged with the treatment and achieved a response similar to the first period, resulting in both patients attaining transfusion independence. The median age was 41, and 44% (21/48) were females. In this sample, 38% (18/48) were splenectomized. Sixteen out of 48 had a β0/β0 genotype, and one had HbE/β-thalassemia. Overall, 37/48 (77%) patients had a TB >15 units in the 24 weeks prior to treatment initiation. Among these, 12 patients had a TB ranging from 20 to 24 units. The median treatment period was 48 weeks (12–172), and 39 out of 48 (81%) received treatment for at least 24 weeks. Seventeen out of 48 (35%) were responders (Figure S1), of whom 10/48 (21%) showed a TB reduction ≥33% in weeks 13–24 (the primary endpoint of the phase 3 trial). Eleven out of 17 (65%) patients in the RSP group had a TB reduction of ≥50% for at least one 12-week interval. Four patients became transfusion-independent for at least 12 weeks, of whom three for at least 22 consecutive weeks. TB, expressed as unit/week, did not change in the NR group (0.7 unit/week in the 24 weeks before luspatercept vs. 0.7 unit/week during the treatment, <i>p</i> = 0.95), while it decreased in the RSP group (0.8 unit/week in the 24 weeks before luspatercept vs. 0.5 unit/week during the treatment, <i>p</i> = 0.001). Overall, in our sample, the mean pretreatment pre-transfusion Hb (pt-Hb) was similar in both groups (Figure 1A). The pt-Hb increased between pre- and during-treatment in both NR (9.1 ± 0.5 g/dL pretreatment vs. 9.3 ± 0.4 g/dL under treatment, <i>p</i> = 0.03) and RSP (from 9.2 ± 0.6 to 9.5 ± 0.5 g/dL, <i>p</i> = 0.025) groups.</p><p>Twenty-one out of 48 (44%) discontinued treatment after at least 12 weeks, with 16 being NR. The reasons for discontinuation are detailed in Table S1.<span><sup>4</sup></span></p><p>The drug was generally well tolerated, and no deaths occurred. The frequency of AEs was similar to those reported in the phase 3 study, and a detailed list can be found in Table S2.</p><p>RSP and NR did not differ in TB during the 24 weeks before treatment initiation when evaluating the number of transfused units, even though iron intake was slightly higher in the NR (0.32 ± 0.07 vs. 0.27 ± 0.08 mg/kg/day, <i>p</i> = 0.09). Patients were overall well chelated. When comparing the two groups, RSP showed higher liver iron concentration (LIC), in a range considered borderline and in the lower range of optimal chelation therapy. As shown in Figure S2 after 24 weeks of treatment, we observed a greater increase in reticulocyte count in RSP than in NR (0.2, 0.0–1.1 × 10<sup>12</sup>/L vs. 0.1, 0.0–0.5 × 10<sup>12</sup>/L, <i>p</i> = 0.04).</p><p>Fetal hemoglobin (HbF) at baseline was the only clinically relevant hematological parameter significantly different between RSP and NR. Also, the baseline absolute value of HbF (g/dL) provided the best discrimination between RSP and NR after a receiver operating characteristic (ROC) analysis, with an area under the curve (AUC) of 0.82 (95% confidence interval [CI] 0.68–0.96; Figure 1B). Baseline values of reticulocytes did not increase the AUC. The statistical cut-off point of the ROC curve was identified for a baseline absolute value of HbF of 0.6 g/dL, corresponding to a negative predictive value (NPV) of 92% (95% CI: 74–99, 23 NR out of 25 with HbF <0.6 g/dL).</p><p>An increase in HbF was also observed in both groups; however, the RSP reached higher values than the NR at 24 weeks, 21.1% (4.2–59.1; corresponding to the median absolute HbF of 2.1 g/dL) versus 7.6% (3.2–37.1; corresponding to the median absolute value of HbF of 0.7 g/dL), respectively (<i>p</i> = 0.0001). In the random-intercept linear regression analysis, the best fit was achieved using a cubic model to describe the trend of HbF increase over time. Both RSP and NR exhibited a similar pattern, with an initial increase in levels that reached a plateau around week 16 (Figure 1C).</p><p>Currently, data on luspatercept safety, efficacy, and potential predictors of response primarily come from clinical trials.<span><sup>1, 2, 5</sup></span> Here, we present findings from the largest published real-world cohort of TDT patients treated with luspatercept. Our study explores predictors of response, which are essential for clinicians to assess the risk–benefit profile when considering prescribing this agent. In our study, the response rate was similar to that of the phase 3 trial when evaluating weeks 13–24, namely the primary endpoint of the BELIEVE study. However, the overall response rate over any 12-week interval in our cohort was lower (35% vs. about 70% in the phase 3 trial). This difference could be attributed to the shorter median treatment duration in our study (48 vs. 64 weeks). Reasons for this include adherence to regulatory guidelines for drug prescription and the patients' involvement in treatment decisions in real life. This can affect, for example, the identification of late-responders.<span><sup>6</sup></span> In our sample, most patients completed 24 weeks of treatment, which is a reasonable time for expecting a response.<span><sup>7</sup></span> Other differences emerged when comparing our data to those of the trial. Our patients were more heavily transfused, with 77% having a TB >15 units of pRBC in the 24 weeks before the initiation of luspatercept, compared to 43.5% in the BELIEVE trial. Interestingly, we have identified baseline HbF levels as a predictor of response to treatment. A baseline HbF <0.6 g/dL was associated with a lower likelihood of response to luspatercept (8% of RSP having baseline HbF <0.6 g/dL). Conversely, TDT patients who have a baseline HbF of 0.6 g/dL or higher exhibit a high likelihood of responding to treatment (64% of patients with HbF >0.6 g/dL were RSP). Of note, those with a baseline HbF <0.6 g/dL did not have a higher TB before luspatercept (median TB in the HbF <0.6 g/dL group: 19 units vs. median TB in the HbF >0.6 g/dL group: 17 units; <i>p</i> = 0.11). We do not recommend excluding a priori patients with a baseline value of HbF <0.6 g/dL from treatment. Rather, we suggest carefully considering initiating luspatercept in these patients, especially in the presence of an uncertain risk–benefit ratio (e.g., a risk factor for an AE). Interestingly, regardless of the baseline level, all patients treated with luspatercept experienced an increase in HbF. However, responders showed a higher increase in absolute HbF values compared to NRs.</p><p>Additionally, the presence of severe genotypes did not prevent a response, as one-fourth of RSP had a β0/β0 genotype.</p><p>Some limitations in this study should be acknowledged. First, the sample size is limited given the rarity of the disease and the recent approval of luspatercept in Italy. Second, the observation period in this study was shorter than that of the Phase 3 BELIEVE trial.</p><p>In conclusion, this study confirms that the effectiveness of luspatercept in a real-world cohort of TDT patients is similar to the phase 3 clinical trial and identifies HbF at baseline and its trend in the first few weeks of treatment as a predictive marker of response. Further studies are needed to elucidate the differences in baseline HbF levels between responders and NRs.</p><p>IM, VB, and DLP conceived and designed the study. GA, SE, SL, MF, NS, VB, DM, IM, and DLP collected the data. DC, DLP, IM, VB, NS, and NC analyzed the data. VB, RB-F, CC, NC, LD, FG, and MAI performed the experiments. IM, DLP, VB, and RB-F wrote the paper. All the authors revised the paper.</p><p>This work was partially supported by the Italian Ministry of Health and Fondazione IRCCS Ca' Granda (RC_2022/23), the funding grant of the Società Italiana di Medicina Interna (SIMI), the grant PRIN2022—project number 2022L5YWZT.</p><p>MDC has been or is a current consultant for Novartis, Celgene Corp, Bristol Myers Squibb, Vifor Pharma, and Ionis Pharmaceuticals and has received research funding from Novartis, Celgene Corp, Bristol Myers Squibb, La Jolla Pharmaceutical Company, Roche, Protagonist Therapeutics, and CRISPR Therapeutics. IM is a scientific advisory board member of Bristol Myers Squibb and Sanofi and received honoraria for lectures from Bristol Myers Squibb and Sanofi and research support from Sanofi.</p><p>Informed consent was obtained from all individual participants included in the study.</p>\",\"PeriodicalId\":7724,\"journal\":{\"name\":\"American Journal of Hematology\",\"volume\":\"99 12\",\"pages\":\"2395-2398\"},\"PeriodicalIF\":10.1000,\"publicationDate\":\"2024-09-12\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/ajh.27474\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"American Journal of Hematology\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/ajh.27474\",\"RegionNum\":1,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"HEMATOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"American Journal of Hematology","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/ajh.27474","RegionNum":1,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"HEMATOLOGY","Score":null,"Total":0}
Real-world efficacy and safety of luspatercept and predictive factors of response in patients with transfusion-dependent β-thalassemia
Luspatercept is the first erythropoiesis-modulating agent approved by the Food and Drug Administration (FDA) and the European Medicines Agency (EMA) for treating anemia in adult transfusion-dependent β-thalassemia (TDT) patients. As observed in clinical trials1 and real-life experience,2 response to luspatercept in TDT is heterogeneous. It can range from patients who do not respond to those who become transfusion-independent. So far, no predictors of response have been identified. However, the definition of the different profiles and predictors of response is necessary for optimizing treatment allocation, limiting costs, and increasing sustainability. The ELEMENT study is an observational prospective cohort study that enrolled adult TDT patients regularly followed at Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico in Milan (Italy) treated with luspatercept. Luspatercept was administered, according to the indications of the Italian Regulatory Agency, subcutaneously at a starting dose of 1.0 mg/kg every 3 weeks and was increased to 1.25 mg/kg after dose 3 if a clinical response was not achieved.3 The drug was discontinued according to the Italian Regulatory Agency indications, namely, if the patient does not achieve transfusion reduction (amount not specified) after three doses at the maximum dosage or in the presence of unacceptable toxicity. Treatment response was assessed by comparing the transfusion burden (TB) during any 12-week treatment period with that in the 24 weeks before treatment. Responders (RSP) were defined as individuals who have a reduction of the TB ≥33% in any 12 weeks of treatment, while those with a TB reduction <33% were considered “non-responders” (NR). Moreover, as in the phase 3 trial, transfusion independence was defined as a transfusion-free period of at least 8 weeks.1 In this study, TB was also expressed as the number of units of packed red blood cells (pRBC) per week (unit/week) by dividing the number of units transfused in a period by the number of weeks evaluated.
Between January 1, 2021, and May 31, 2024, 56 TDT patients received at least one dose of luspatercept after drug authorization. At the start of the study, treatment was offered to TDT patients with high TB, iron overload demonstrated by T2* magnetic resonance imaging (MRI), or other clinically relevant conditions that might benefit from TB reduction. Subsequently, the treatment was made available to all the patients who met the Italian regulatory agency criteria and were willing to receive the drug. The decision to initiate treatment was always discussed between the physician and the patient.
Seven patients discontinued the drug before completing at least 12 weeks. The reasons for discontinuation are detailed in Table S1 (early drug discontinuation group). At the time of the analysis, one patient had not yet completed 12 weeks of treatment.
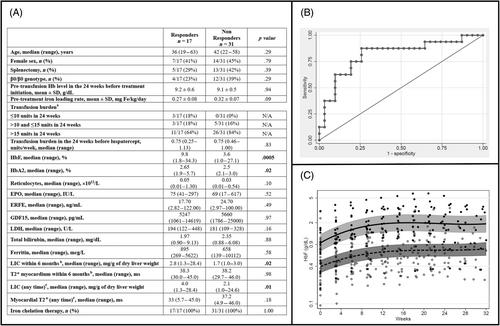
Data from the 48 patients who received luspatercept for at least 12 weeks were included, and their characteristics at enrollment are presented in Figure 1A. Out of 48 patients, two paused their treatment temporarily, one due to a personal decision and the other due to an adverse event (AE). They were later re-challenged with the treatment and achieved a response similar to the first period, resulting in both patients attaining transfusion independence. The median age was 41, and 44% (21/48) were females. In this sample, 38% (18/48) were splenectomized. Sixteen out of 48 had a β0/β0 genotype, and one had HbE/β-thalassemia. Overall, 37/48 (77%) patients had a TB >15 units in the 24 weeks prior to treatment initiation. Among these, 12 patients had a TB ranging from 20 to 24 units. The median treatment period was 48 weeks (12–172), and 39 out of 48 (81%) received treatment for at least 24 weeks. Seventeen out of 48 (35%) were responders (Figure S1), of whom 10/48 (21%) showed a TB reduction ≥33% in weeks 13–24 (the primary endpoint of the phase 3 trial). Eleven out of 17 (65%) patients in the RSP group had a TB reduction of ≥50% for at least one 12-week interval. Four patients became transfusion-independent for at least 12 weeks, of whom three for at least 22 consecutive weeks. TB, expressed as unit/week, did not change in the NR group (0.7 unit/week in the 24 weeks before luspatercept vs. 0.7 unit/week during the treatment, p = 0.95), while it decreased in the RSP group (0.8 unit/week in the 24 weeks before luspatercept vs. 0.5 unit/week during the treatment, p = 0.001). Overall, in our sample, the mean pretreatment pre-transfusion Hb (pt-Hb) was similar in both groups (Figure 1A). The pt-Hb increased between pre- and during-treatment in both NR (9.1 ± 0.5 g/dL pretreatment vs. 9.3 ± 0.4 g/dL under treatment, p = 0.03) and RSP (from 9.2 ± 0.6 to 9.5 ± 0.5 g/dL, p = 0.025) groups.
Twenty-one out of 48 (44%) discontinued treatment after at least 12 weeks, with 16 being NR. The reasons for discontinuation are detailed in Table S1.4
The drug was generally well tolerated, and no deaths occurred. The frequency of AEs was similar to those reported in the phase 3 study, and a detailed list can be found in Table S2.
RSP and NR did not differ in TB during the 24 weeks before treatment initiation when evaluating the number of transfused units, even though iron intake was slightly higher in the NR (0.32 ± 0.07 vs. 0.27 ± 0.08 mg/kg/day, p = 0.09). Patients were overall well chelated. When comparing the two groups, RSP showed higher liver iron concentration (LIC), in a range considered borderline and in the lower range of optimal chelation therapy. As shown in Figure S2 after 24 weeks of treatment, we observed a greater increase in reticulocyte count in RSP than in NR (0.2, 0.0–1.1 × 1012/L vs. 0.1, 0.0–0.5 × 1012/L, p = 0.04).
Fetal hemoglobin (HbF) at baseline was the only clinically relevant hematological parameter significantly different between RSP and NR. Also, the baseline absolute value of HbF (g/dL) provided the best discrimination between RSP and NR after a receiver operating characteristic (ROC) analysis, with an area under the curve (AUC) of 0.82 (95% confidence interval [CI] 0.68–0.96; Figure 1B). Baseline values of reticulocytes did not increase the AUC. The statistical cut-off point of the ROC curve was identified for a baseline absolute value of HbF of 0.6 g/dL, corresponding to a negative predictive value (NPV) of 92% (95% CI: 74–99, 23 NR out of 25 with HbF <0.6 g/dL).
An increase in HbF was also observed in both groups; however, the RSP reached higher values than the NR at 24 weeks, 21.1% (4.2–59.1; corresponding to the median absolute HbF of 2.1 g/dL) versus 7.6% (3.2–37.1; corresponding to the median absolute value of HbF of 0.7 g/dL), respectively (p = 0.0001). In the random-intercept linear regression analysis, the best fit was achieved using a cubic model to describe the trend of HbF increase over time. Both RSP and NR exhibited a similar pattern, with an initial increase in levels that reached a plateau around week 16 (Figure 1C).
Currently, data on luspatercept safety, efficacy, and potential predictors of response primarily come from clinical trials.1, 2, 5 Here, we present findings from the largest published real-world cohort of TDT patients treated with luspatercept. Our study explores predictors of response, which are essential for clinicians to assess the risk–benefit profile when considering prescribing this agent. In our study, the response rate was similar to that of the phase 3 trial when evaluating weeks 13–24, namely the primary endpoint of the BELIEVE study. However, the overall response rate over any 12-week interval in our cohort was lower (35% vs. about 70% in the phase 3 trial). This difference could be attributed to the shorter median treatment duration in our study (48 vs. 64 weeks). Reasons for this include adherence to regulatory guidelines for drug prescription and the patients' involvement in treatment decisions in real life. This can affect, for example, the identification of late-responders.6 In our sample, most patients completed 24 weeks of treatment, which is a reasonable time for expecting a response.7 Other differences emerged when comparing our data to those of the trial. Our patients were more heavily transfused, with 77% having a TB >15 units of pRBC in the 24 weeks before the initiation of luspatercept, compared to 43.5% in the BELIEVE trial. Interestingly, we have identified baseline HbF levels as a predictor of response to treatment. A baseline HbF <0.6 g/dL was associated with a lower likelihood of response to luspatercept (8% of RSP having baseline HbF <0.6 g/dL). Conversely, TDT patients who have a baseline HbF of 0.6 g/dL or higher exhibit a high likelihood of responding to treatment (64% of patients with HbF >0.6 g/dL were RSP). Of note, those with a baseline HbF <0.6 g/dL did not have a higher TB before luspatercept (median TB in the HbF <0.6 g/dL group: 19 units vs. median TB in the HbF >0.6 g/dL group: 17 units; p = 0.11). We do not recommend excluding a priori patients with a baseline value of HbF <0.6 g/dL from treatment. Rather, we suggest carefully considering initiating luspatercept in these patients, especially in the presence of an uncertain risk–benefit ratio (e.g., a risk factor for an AE). Interestingly, regardless of the baseline level, all patients treated with luspatercept experienced an increase in HbF. However, responders showed a higher increase in absolute HbF values compared to NRs.
Additionally, the presence of severe genotypes did not prevent a response, as one-fourth of RSP had a β0/β0 genotype.
Some limitations in this study should be acknowledged. First, the sample size is limited given the rarity of the disease and the recent approval of luspatercept in Italy. Second, the observation period in this study was shorter than that of the Phase 3 BELIEVE trial.
In conclusion, this study confirms that the effectiveness of luspatercept in a real-world cohort of TDT patients is similar to the phase 3 clinical trial and identifies HbF at baseline and its trend in the first few weeks of treatment as a predictive marker of response. Further studies are needed to elucidate the differences in baseline HbF levels between responders and NRs.
IM, VB, and DLP conceived and designed the study. GA, SE, SL, MF, NS, VB, DM, IM, and DLP collected the data. DC, DLP, IM, VB, NS, and NC analyzed the data. VB, RB-F, CC, NC, LD, FG, and MAI performed the experiments. IM, DLP, VB, and RB-F wrote the paper. All the authors revised the paper.
This work was partially supported by the Italian Ministry of Health and Fondazione IRCCS Ca' Granda (RC_2022/23), the funding grant of the Società Italiana di Medicina Interna (SIMI), the grant PRIN2022—project number 2022L5YWZT.
MDC has been or is a current consultant for Novartis, Celgene Corp, Bristol Myers Squibb, Vifor Pharma, and Ionis Pharmaceuticals and has received research funding from Novartis, Celgene Corp, Bristol Myers Squibb, La Jolla Pharmaceutical Company, Roche, Protagonist Therapeutics, and CRISPR Therapeutics. IM is a scientific advisory board member of Bristol Myers Squibb and Sanofi and received honoraria for lectures from Bristol Myers Squibb and Sanofi and research support from Sanofi.
Informed consent was obtained from all individual participants included in the study.
期刊介绍:
The American Journal of Hematology offers extensive coverage of experimental and clinical aspects of blood diseases in humans and animal models. The journal publishes original contributions in both non-malignant and malignant hematological diseases, encompassing clinical and basic studies in areas such as hemostasis, thrombosis, immunology, blood banking, and stem cell biology. Clinical translational reports highlighting innovative therapeutic approaches for the diagnosis and treatment of hematological diseases are actively encouraged.The American Journal of Hematology features regular original laboratory and clinical research articles, brief research reports, critical reviews, images in hematology, as well as letters and correspondence.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


