{"title":"基于序列的甲型流感病毒亚型分类法的前景。","authors":"Art F Y Poon","doi":"10.1093/ve/veae064","DOIUrl":null,"url":null,"abstract":"<p><p>Hemagglutinin (HA) and neuraminidase (NA) proteins are the primary antigenic targets of influenza A virus (IAV) infections. IAV infections are generally classified into subtypes of HA and NA proteins, e.g. H3N2. Most of the known subtypes were originally defined by a lack of antibody cross-reactivity. However, genetic sequencing has played an increasingly important role in characterizing the evolving diversity of IAV. Novel subtypes have recently been described solely by their genetic sequences, and IAV infections are routinely subtyped by molecular assays, or the comparison of sequences to references. In this study, I carry out a comparative analysis of all available IAV protein sequences in the Genbank database (over 1.1 million, reduced to 272,292 unique sequences prior to phylogenetic reconstruction) to determine whether the serologically defined subtypes can be reproduced with sequence-based criteria. I show that a robust genetic taxonomy of HA and NA subtypes can be obtained using a simple clustering method, namely, by progressively partitioning the phylogeny on its longest internal branches. However, this taxonomy also requires some amendments to the current nomenclature. For example, two IAV isolates from bats previously characterized as a divergent lineage of H9N2 should be separated into their own subtype. With the exception of these small and highly divergent lineages, the phylogenies relating each of the other six genomic segments do not support partitions into major subtypes.</p>","PeriodicalId":56026,"journal":{"name":"Virus Evolution","volume":"10 1","pages":"veae064"},"PeriodicalIF":4.0000,"publicationDate":"2024-08-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11378807/pdf/","citationCount":"0","resultStr":"{\"title\":\"Prospects for a sequence-based taxonomy of influenza A virus subtypes.\",\"authors\":\"Art F Y Poon\",\"doi\":\"10.1093/ve/veae064\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Hemagglutinin (HA) and neuraminidase (NA) proteins are the primary antigenic targets of influenza A virus (IAV) infections. IAV infections are generally classified into subtypes of HA and NA proteins, e.g. H3N2. Most of the known subtypes were originally defined by a lack of antibody cross-reactivity. However, genetic sequencing has played an increasingly important role in characterizing the evolving diversity of IAV. Novel subtypes have recently been described solely by their genetic sequences, and IAV infections are routinely subtyped by molecular assays, or the comparison of sequences to references. In this study, I carry out a comparative analysis of all available IAV protein sequences in the Genbank database (over 1.1 million, reduced to 272,292 unique sequences prior to phylogenetic reconstruction) to determine whether the serologically defined subtypes can be reproduced with sequence-based criteria. I show that a robust genetic taxonomy of HA and NA subtypes can be obtained using a simple clustering method, namely, by progressively partitioning the phylogeny on its longest internal branches. However, this taxonomy also requires some amendments to the current nomenclature. For example, two IAV isolates from bats previously characterized as a divergent lineage of H9N2 should be separated into their own subtype. With the exception of these small and highly divergent lineages, the phylogenies relating each of the other six genomic segments do not support partitions into major subtypes.</p>\",\"PeriodicalId\":56026,\"journal\":{\"name\":\"Virus Evolution\",\"volume\":\"10 1\",\"pages\":\"veae064\"},\"PeriodicalIF\":4.0000,\"publicationDate\":\"2024-08-17\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11378807/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Virus Evolution\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1093/ve/veae064\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q1\",\"JCRName\":\"VIROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Virus Evolution","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1093/ve/veae064","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/1/1 0:00:00","PubModel":"eCollection","JCR":"Q1","JCRName":"VIROLOGY","Score":null,"Total":0}
引用次数: 0
摘要
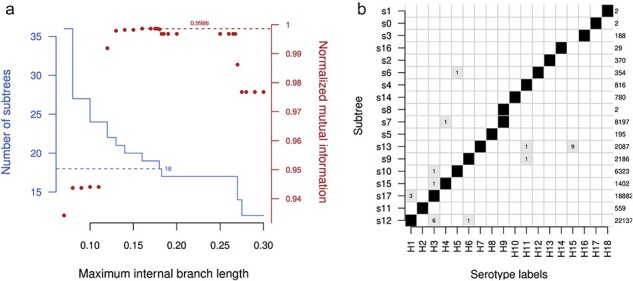
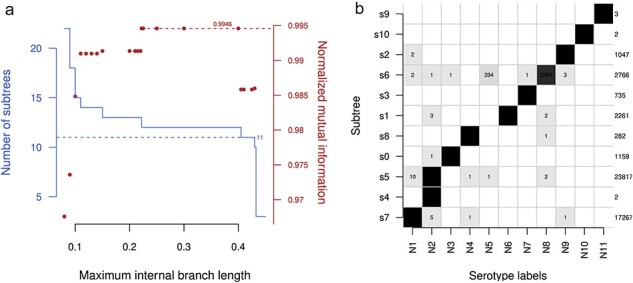
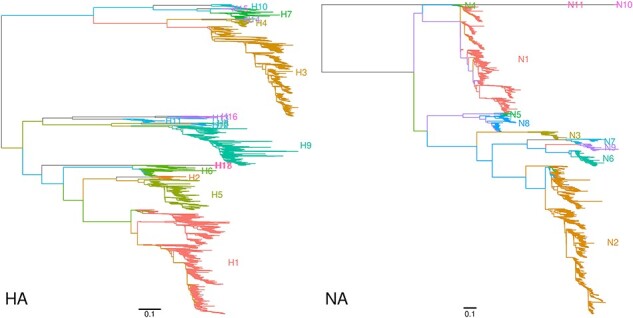
血凝素(HA)和神经氨酸酶(NA)蛋白是甲型流感病毒(IAV)感染的主要抗原目标。IAV 感染一般分为 HA 和 NA 蛋白亚型,如 H3N2。大多数已知亚型最初是通过缺乏抗体交叉反应来定义的。然而,基因测序在描述 IAV 不断演变的多样性方面发挥着越来越重要的作用。最近,一些新的亚型仅通过其基因序列就被描述出来,而 IAV 感染通常是通过分子检测或将序列与参考文献进行比较来确定亚型的。在本研究中,我对 Genbank 数据库中所有可用的 IAV 蛋白序列(超过 110 万个,在系统发育重建前已减少到 272 292 个唯一序列)进行了比较分析,以确定血清学定义的亚型是否可以通过基于序列的标准再现。我的研究表明,使用简单的聚类方法,即在最长的内部分支上逐步划分系统发育,就能获得稳健的 HA 和 NA 亚型遗传分类法。不过,这种分类法也需要对目前的命名法进行一些修改。例如,以前被定性为 H9N2 分歧系的两个蝙蝠 IAV 分离物应被分离成各自的亚型。除了这些高度分化的小系外,与其他六个基因组片段相关的系统发育并不支持将其划分为主要亚型。
Prospects for a sequence-based taxonomy of influenza A virus subtypes.
Hemagglutinin (HA) and neuraminidase (NA) proteins are the primary antigenic targets of influenza A virus (IAV) infections. IAV infections are generally classified into subtypes of HA and NA proteins, e.g. H3N2. Most of the known subtypes were originally defined by a lack of antibody cross-reactivity. However, genetic sequencing has played an increasingly important role in characterizing the evolving diversity of IAV. Novel subtypes have recently been described solely by their genetic sequences, and IAV infections are routinely subtyped by molecular assays, or the comparison of sequences to references. In this study, I carry out a comparative analysis of all available IAV protein sequences in the Genbank database (over 1.1 million, reduced to 272,292 unique sequences prior to phylogenetic reconstruction) to determine whether the serologically defined subtypes can be reproduced with sequence-based criteria. I show that a robust genetic taxonomy of HA and NA subtypes can be obtained using a simple clustering method, namely, by progressively partitioning the phylogeny on its longest internal branches. However, this taxonomy also requires some amendments to the current nomenclature. For example, two IAV isolates from bats previously characterized as a divergent lineage of H9N2 should be separated into their own subtype. With the exception of these small and highly divergent lineages, the phylogenies relating each of the other six genomic segments do not support partitions into major subtypes.
期刊介绍:
Virus Evolution is a new Open Access journal focusing on the long-term evolution of viruses, viruses as a model system for studying evolutionary processes, viral molecular epidemiology and environmental virology.
The aim of the journal is to provide a forum for original research papers, reviews, commentaries and a venue for in-depth discussion on the topics relevant to virus evolution.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


