{"title":"关于狄拉克半金属 AMgBi(A=K、Rb、Cs)物理性质的 Ab-Initio 计算","authors":"Bahadır Salmankurt","doi":"10.1016/j.jssc.2024.125000","DOIUrl":null,"url":null,"abstract":"<div><p>This study presents a comprehensive first-principles investigation of the structural, mechanical, vibrational, thermodynamic and electronic properties of AMgBi (A = K, Rb, Cs) compounds using Density Functional Theory. Also, the effect of substituting alkali atoms on the physical properties has been discussed. The tetragonal PbClF-type structure has been confirmed by the analysis and the results revealed good agreement between calculated and experimental lattice parameters for KMgBi. The mechanical properties have been investigated for the first time for RbMgBi and CsMgBi. The mechanical stability of the materials in the ground state has been confirmed through the use of obtained elastic constants. Furthermore, derived parameters from elastic constants such as bulk modulus, shear modulus, and Poisson's ratio indicated that the materials are brittle and exhibited anisotropic mechanical behavior due to their layered structure. This study conducts a detailed analysis of phonon modes, explores their connections to thermal and elastic properties, visualizes the movements of phonon modes at the gamma point, determines Born effective charges, and discusses LO/TO splitting, which is for the first time for RbMgBi and CsMgBi. Phonon dispersion calculations confirmed the dynamical stability of the compounds and revealed the presence of phonon band gaps, supporting their quasi-two-dimensional nature. Investigation of thermodynamic properties using the quasi-harmonic approximation has shown the temperature dependence of internal energy, Helmholtz free energy, specific heat, and entropy for the first time for all compounds. The materials exhibited relatively low thermal conductivity, following the order KMgBi > RbMgBi > CsMgBi. The calculated Grüneisen parameter values were found to be 1.42, 1.44, and 1.53 for KMgBi, RbMgBi, and CsMgBi, respectively, suggesting relatively weak anharmonicity within the materials.</p></div>","PeriodicalId":378,"journal":{"name":"Journal of Solid State Chemistry","volume":"340 ","pages":"Article 125000"},"PeriodicalIF":3.2000,"publicationDate":"2024-09-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Ab-Initio calculations on physical properties of Dirac semimetal AMgBi (A=K, Rb, Cs)\",\"authors\":\"Bahadır Salmankurt\",\"doi\":\"10.1016/j.jssc.2024.125000\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>This study presents a comprehensive first-principles investigation of the structural, mechanical, vibrational, thermodynamic and electronic properties of AMgBi (A = K, Rb, Cs) compounds using Density Functional Theory. Also, the effect of substituting alkali atoms on the physical properties has been discussed. The tetragonal PbClF-type structure has been confirmed by the analysis and the results revealed good agreement between calculated and experimental lattice parameters for KMgBi. The mechanical properties have been investigated for the first time for RbMgBi and CsMgBi. The mechanical stability of the materials in the ground state has been confirmed through the use of obtained elastic constants. Furthermore, derived parameters from elastic constants such as bulk modulus, shear modulus, and Poisson's ratio indicated that the materials are brittle and exhibited anisotropic mechanical behavior due to their layered structure. This study conducts a detailed analysis of phonon modes, explores their connections to thermal and elastic properties, visualizes the movements of phonon modes at the gamma point, determines Born effective charges, and discusses LO/TO splitting, which is for the first time for RbMgBi and CsMgBi. Phonon dispersion calculations confirmed the dynamical stability of the compounds and revealed the presence of phonon band gaps, supporting their quasi-two-dimensional nature. Investigation of thermodynamic properties using the quasi-harmonic approximation has shown the temperature dependence of internal energy, Helmholtz free energy, specific heat, and entropy for the first time for all compounds. The materials exhibited relatively low thermal conductivity, following the order KMgBi > RbMgBi > CsMgBi. The calculated Grüneisen parameter values were found to be 1.42, 1.44, and 1.53 for KMgBi, RbMgBi, and CsMgBi, respectively, suggesting relatively weak anharmonicity within the materials.</p></div>\",\"PeriodicalId\":378,\"journal\":{\"name\":\"Journal of Solid State Chemistry\",\"volume\":\"340 \",\"pages\":\"Article 125000\"},\"PeriodicalIF\":3.2000,\"publicationDate\":\"2024-09-04\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Solid State Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S0022459624004547\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, INORGANIC & NUCLEAR\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Solid State Chemistry","FirstCategoryId":"92","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0022459624004547","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, INORGANIC & NUCLEAR","Score":null,"Total":0}
Ab-Initio calculations on physical properties of Dirac semimetal AMgBi (A=K, Rb, Cs)
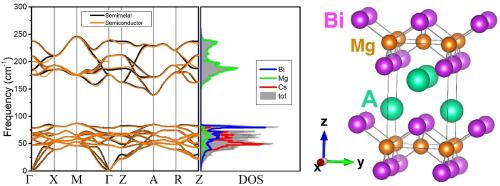
This study presents a comprehensive first-principles investigation of the structural, mechanical, vibrational, thermodynamic and electronic properties of AMgBi (A = K, Rb, Cs) compounds using Density Functional Theory. Also, the effect of substituting alkali atoms on the physical properties has been discussed. The tetragonal PbClF-type structure has been confirmed by the analysis and the results revealed good agreement between calculated and experimental lattice parameters for KMgBi. The mechanical properties have been investigated for the first time for RbMgBi and CsMgBi. The mechanical stability of the materials in the ground state has been confirmed through the use of obtained elastic constants. Furthermore, derived parameters from elastic constants such as bulk modulus, shear modulus, and Poisson's ratio indicated that the materials are brittle and exhibited anisotropic mechanical behavior due to their layered structure. This study conducts a detailed analysis of phonon modes, explores their connections to thermal and elastic properties, visualizes the movements of phonon modes at the gamma point, determines Born effective charges, and discusses LO/TO splitting, which is for the first time for RbMgBi and CsMgBi. Phonon dispersion calculations confirmed the dynamical stability of the compounds and revealed the presence of phonon band gaps, supporting their quasi-two-dimensional nature. Investigation of thermodynamic properties using the quasi-harmonic approximation has shown the temperature dependence of internal energy, Helmholtz free energy, specific heat, and entropy for the first time for all compounds. The materials exhibited relatively low thermal conductivity, following the order KMgBi > RbMgBi > CsMgBi. The calculated Grüneisen parameter values were found to be 1.42, 1.44, and 1.53 for KMgBi, RbMgBi, and CsMgBi, respectively, suggesting relatively weak anharmonicity within the materials.
期刊介绍:
Covering major developments in the field of solid state chemistry and related areas such as ceramics and amorphous materials, the Journal of Solid State Chemistry features studies of chemical, structural, thermodynamic, electronic, magnetic, and optical properties and processes in solids.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


