铁硫簇组装基因 IBA57 突变的表型谱:在自然病史稳定的中国患者中发现的 c.286 T > C 热点突变。
IF 2.6
3区 生物学
Q2 GENETICS & HEREDITY
引用次数: 0
摘要
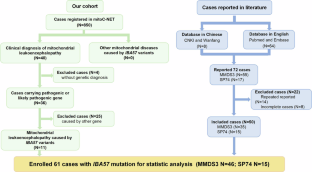
IBA57 基因突变会破坏铁硫簇的成熟,从而导致一种罕见的线粒体疾病。临床表现从新生儿致死到儿童期发病的痉挛性截瘫不等,但其种族异质性和自然史仍不清楚,因此有必要进一步探讨。本研究旨在通过分析不同的临床表现,阐明 IBA57 基因突变的基因型与表型之间的相关性。我们报告了 11 例中国患者,并纳入了文献报道的病例,共计 61 例患者参与了分析。我们收集了临床、神经影像学、遗传和疾病进展信息。其中,46 例表现为多线粒体功能障碍综合征 3(MMDS3),58.7% 来自中国人群。根据病程,我们提出了三种临床亚型:新生儿亚型、婴儿亚型和儿童亚型。新生儿病例在发病时普遍表现为肌张力低下和呼吸窘迫,在三个月内死亡。大多数婴儿和儿童病例表现为发育倒退和运动功能受损。腔隙性白质脑病是 MMDS3 患者典型的神经影像学表现。据报道,85.2%的中国患者存在c.286 T > C突变。与非中国组相比,中国患者的死亡率明显降低(P = 0.002),5年生存率超过90%,表明疾病进展相对稳定。来自三个家族的15例患者表现为痉挛性截瘫74表型,发病前发育正常,常见临床表现包括痉挛性截瘫(14/15)、视力障碍(10/13)和周围神经病变(9/13)。总之,本研究指出了中国人的一个热点突变,并分析了不同临床亚型的疾病进展。本文章由计算机程序翻译,如有差异,请以英文原文为准。

Phenotypic spectrum of iron-sulfur cluster assembly gene IBA57 mutations: c.286 T > C identified as a hotspot mutation in Chinese patients with a stable natural history
Mutations in IBA57 disrupt iron-sulfur clusters maturation, causing a rare mitochondrial disease. Clinical manifestations vary from neonatal lethality to childhood-onset spastic paraparesis, yet the ethnic heterogeneity and natural history remain unclear, necessitating further exploration. This study aimed to delineate the genotype-phenotype correlation of IBA57 mutations by analyzing diverse clinical presentations. We report 11 Chinese patients and include literature-reported cases, totaling 61 patients enrolled for analysis. Clinical, neuroimaging, genetic, and disease progression information were collected. Among these, 46 presented as multiple mitochondrial dysfunctions syndrome 3 (MMDS3), with 58.7% originating from Chinese population. Based on disease course, we propose three clinical subtypes: neonatal, infant and childhood subtypes. Neonatal cases universally displayed hypotonia and respiratory distress at presentation, deceased within three months. Most infancy and childhood cases exhibited developmental regression and impaired motor function. Cavitating leukoencephalopathy was a typical neuroimaging finding in MMDS3 patients. The c.286 T > C mutation was reported in 85.2% of Chinese patients. A significantly lower mortality rate was observed compared to the non-Chinese group (P = 0.002), with a survival rate exceeding 90% at 5 years, indicating a relatively stable disease progression. Fifteen cases from three families manifested the spastic paraplegia 74 phenotype, demonstrating normal development before onset, with common clinical manifestations including spastic paraplegia (14/15), visual impairment (10/13), and peripheral neuropathy (9/13). In conclusion, this study indicates a hotspot mutation in Chinese and analyses the disease progression with different clinical subtypes.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Journal of Human Genetics
生物-遗传学
CiteScore
7.20
自引率
0.00%
发文量
101
审稿时长
4-8 weeks
期刊介绍:
The Journal of Human Genetics is an international journal publishing articles on human genetics, including medical genetics and human genome analysis. It covers all aspects of human genetics, including molecular genetics, clinical genetics, behavioral genetics, immunogenetics, pharmacogenomics, population genetics, functional genomics, epigenetics, genetic counseling and gene therapy.
Articles on the following areas are especially welcome: genetic factors of monogenic and complex disorders, genome-wide association studies, genetic epidemiology, cancer genetics, personal genomics, genotype-phenotype relationships and genome diversity.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


