{"title":"从韩国成年人中分离出的艾希病毒的全基因组测序和基因组特征。","authors":"Seoyoung Woo, Md Iqbal Hossain, Soontag Jung, Daseul Yeo, Danbi Yoon, Seongwon Hwang, Hee-Jung Do, Seong-il Eyun, Changsun Choi","doi":"10.1002/jmv.29902","DOIUrl":null,"url":null,"abstract":"<p>The whole-genome sequence (WGS) analysis of Aichivirus (AiV) identified in Korea was performed in this study. Using Sanger and Nanopore sequencing, the 8228-nucleotide-long genomic sequence of AiV (OQ121963) was determined and confirmed to belong to genotype A. The full-length genome of OQ121963 consisted of a 7296 nt open reading frame (ORF) that encodes a single polyprotein, and 5′ UTR (676 nt) and 3′ UTR (256 nt) at 5′ and 3′ ends, respectively. The ORF consisted of leader protein (L), structural protein P1 (VP0, VP1, and VP3), and nonstructural protein P2 (2A, 2B, and 2C) and P3 (3A, 3B, 3C, and 3D). The secondary structure analysis of the 5′ UTR identified only stem-loop C (SL–C) and not SL–A and SL–B. The variable region of the AiV genome was analyzed by MegAlign Pro and reconfirmed by SimPlot analysis using 16 AiV whole genomes known to date. Among the entire regions, structural protein region P1 showed the lowest amino acid identity (96.07%) with reference sequence AB040749 (originated in Japan; genotype A), while the highest amino acid identity (98.26%) was confirmed in the 3D region among nonstructural protein region P2 and P3. Moreover, phylogenetic analysis of the WGS of OQ121963 showed the highest homology (96.96%) with JX564249 (originated in Taiwan; genotype A) and lowest homology (90.14%) with DQ028632 (originated in Brazil; genotype B). Therefore, the complete genome characterization of OQ121963 and phylogenetic analysis of the AiV conducted in this study provide useful information allowing to improve diagnostic tools and epidemiological studies of AiVs.</p>","PeriodicalId":16354,"journal":{"name":"Journal of Medical Virology","volume":null,"pages":null},"PeriodicalIF":6.8000,"publicationDate":"2024-09-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jmv.29902","citationCount":"0","resultStr":"{\"title\":\"Whole genome sequencing and genome characterization of Aichivirus isolated from Korean adults\",\"authors\":\"Seoyoung Woo, Md Iqbal Hossain, Soontag Jung, Daseul Yeo, Danbi Yoon, Seongwon Hwang, Hee-Jung Do, Seong-il Eyun, Changsun Choi\",\"doi\":\"10.1002/jmv.29902\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>The whole-genome sequence (WGS) analysis of Aichivirus (AiV) identified in Korea was performed in this study. Using Sanger and Nanopore sequencing, the 8228-nucleotide-long genomic sequence of AiV (OQ121963) was determined and confirmed to belong to genotype A. The full-length genome of OQ121963 consisted of a 7296 nt open reading frame (ORF) that encodes a single polyprotein, and 5′ UTR (676 nt) and 3′ UTR (256 nt) at 5′ and 3′ ends, respectively. The ORF consisted of leader protein (L), structural protein P1 (VP0, VP1, and VP3), and nonstructural protein P2 (2A, 2B, and 2C) and P3 (3A, 3B, 3C, and 3D). The secondary structure analysis of the 5′ UTR identified only stem-loop C (SL–C) and not SL–A and SL–B. The variable region of the AiV genome was analyzed by MegAlign Pro and reconfirmed by SimPlot analysis using 16 AiV whole genomes known to date. Among the entire regions, structural protein region P1 showed the lowest amino acid identity (96.07%) with reference sequence AB040749 (originated in Japan; genotype A), while the highest amino acid identity (98.26%) was confirmed in the 3D region among nonstructural protein region P2 and P3. Moreover, phylogenetic analysis of the WGS of OQ121963 showed the highest homology (96.96%) with JX564249 (originated in Taiwan; genotype A) and lowest homology (90.14%) with DQ028632 (originated in Brazil; genotype B). Therefore, the complete genome characterization of OQ121963 and phylogenetic analysis of the AiV conducted in this study provide useful information allowing to improve diagnostic tools and epidemiological studies of AiVs.</p>\",\"PeriodicalId\":16354,\"journal\":{\"name\":\"Journal of Medical Virology\",\"volume\":null,\"pages\":null},\"PeriodicalIF\":6.8000,\"publicationDate\":\"2024-09-04\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jmv.29902\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Medical Virology\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/jmv.29902\",\"RegionNum\":3,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"VIROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Medical Virology","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jmv.29902","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"VIROLOGY","Score":null,"Total":0}
Whole genome sequencing and genome characterization of Aichivirus isolated from Korean adults
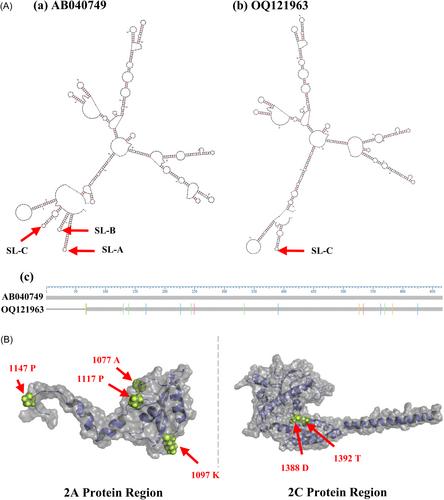
The whole-genome sequence (WGS) analysis of Aichivirus (AiV) identified in Korea was performed in this study. Using Sanger and Nanopore sequencing, the 8228-nucleotide-long genomic sequence of AiV (OQ121963) was determined and confirmed to belong to genotype A. The full-length genome of OQ121963 consisted of a 7296 nt open reading frame (ORF) that encodes a single polyprotein, and 5′ UTR (676 nt) and 3′ UTR (256 nt) at 5′ and 3′ ends, respectively. The ORF consisted of leader protein (L), structural protein P1 (VP0, VP1, and VP3), and nonstructural protein P2 (2A, 2B, and 2C) and P3 (3A, 3B, 3C, and 3D). The secondary structure analysis of the 5′ UTR identified only stem-loop C (SL–C) and not SL–A and SL–B. The variable region of the AiV genome was analyzed by MegAlign Pro and reconfirmed by SimPlot analysis using 16 AiV whole genomes known to date. Among the entire regions, structural protein region P1 showed the lowest amino acid identity (96.07%) with reference sequence AB040749 (originated in Japan; genotype A), while the highest amino acid identity (98.26%) was confirmed in the 3D region among nonstructural protein region P2 and P3. Moreover, phylogenetic analysis of the WGS of OQ121963 showed the highest homology (96.96%) with JX564249 (originated in Taiwan; genotype A) and lowest homology (90.14%) with DQ028632 (originated in Brazil; genotype B). Therefore, the complete genome characterization of OQ121963 and phylogenetic analysis of the AiV conducted in this study provide useful information allowing to improve diagnostic tools and epidemiological studies of AiVs.
期刊介绍:
The Journal of Medical Virology focuses on publishing original scientific papers on both basic and applied research related to viruses that affect humans. The journal publishes reports covering a wide range of topics, including the characterization, diagnosis, epidemiology, immunology, and pathogenesis of human virus infections. It also includes studies on virus morphology, genetics, replication, and interactions with host cells.
The intended readership of the journal includes virologists, microbiologists, immunologists, infectious disease specialists, diagnostic laboratory technologists, epidemiologists, hematologists, and cell biologists.
The Journal of Medical Virology is indexed and abstracted in various databases, including Abstracts in Anthropology (Sage), CABI, AgBiotech News & Information, National Agricultural Library, Biological Abstracts, Embase, Global Health, Web of Science, Veterinary Bulletin, and others.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


