{"title":"通过抑制 NLRC4/IRF1 信号通路,缺失 AIM2 可减轻高纤维脂肪饮食/STZ 诱导的糖尿病小鼠的心脏炎症和肥大。","authors":"Jian-Ping Wu, Cheng Wu, Yuan-Ji Ma, Jian-Bing Zhu, Lei-Lei Ma, Fei-Juan Kong","doi":"10.1007/s12265-024-10556-0","DOIUrl":null,"url":null,"abstract":"<p><p>Absent in melanoma 2(AIM2) exacerbates atherosclerosis by inflammasome assembly. However, AIM2-mediated inflammation in diabetic cardiomyopathy remains incompletely understood. Here we investigate the role of AIM2 in high glucose (HG)- and diabetes-induced inflammatory cardiomyopathy. By RNA-seq, we found that AIM2 were significantly upregulated in HG-induced macrophages, upregulation of AIM2 in cardiac infiltrating macrophages was confirmed in a high-fat diet (HFD)/streptozotocin (STZ)-induceddiabetic mouse model . Therefore, AIM2 knockout mice were constructed. Compared to WT mice, HFD/STZ-induced cardiac hypertrophy and dysfunction were significantly improved in AIM2<sup>-/-</sup> mice, despite no changes in blood glucose and body weight. Further, AIM2 deficiency inhibited cardiac recruitment of M1-macrophages and cytokine production. Mechanistically, AIM2-deficient macrophgaes reduced IL-1β and TNF-α secretion, which impaired the NLRC4/IRF1 signaling in cardiomyocytes, and reduced further recruitment of macrophages, attenuated cardiac inflammation and hypertrophy, these effects were confirmed by silencing IRF1 in WT mice, and significantly reversed by overexpression of IRF1 in AIM2<sup>-/-</sup> mice. Taken together, our findings suggest that AIM2 serves as a novel target for the treatment of diabetic cardiomyopathy.</p>","PeriodicalId":15224,"journal":{"name":"Journal of Cardiovascular Translational Research","volume":" ","pages":"94-109"},"PeriodicalIF":2.5000,"publicationDate":"2025-02-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"AIM2 Deficiency Alleviates Cardiac Inflammation and Hypertrophy in HFD/STZ-Induced Diabetic Mice by Inhibiting the NLRC4/IRF1 Signaling Pathway.\",\"authors\":\"Jian-Ping Wu, Cheng Wu, Yuan-Ji Ma, Jian-Bing Zhu, Lei-Lei Ma, Fei-Juan Kong\",\"doi\":\"10.1007/s12265-024-10556-0\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Absent in melanoma 2(AIM2) exacerbates atherosclerosis by inflammasome assembly. However, AIM2-mediated inflammation in diabetic cardiomyopathy remains incompletely understood. Here we investigate the role of AIM2 in high glucose (HG)- and diabetes-induced inflammatory cardiomyopathy. By RNA-seq, we found that AIM2 were significantly upregulated in HG-induced macrophages, upregulation of AIM2 in cardiac infiltrating macrophages was confirmed in a high-fat diet (HFD)/streptozotocin (STZ)-induceddiabetic mouse model . Therefore, AIM2 knockout mice were constructed. Compared to WT mice, HFD/STZ-induced cardiac hypertrophy and dysfunction were significantly improved in AIM2<sup>-/-</sup> mice, despite no changes in blood glucose and body weight. Further, AIM2 deficiency inhibited cardiac recruitment of M1-macrophages and cytokine production. Mechanistically, AIM2-deficient macrophgaes reduced IL-1β and TNF-α secretion, which impaired the NLRC4/IRF1 signaling in cardiomyocytes, and reduced further recruitment of macrophages, attenuated cardiac inflammation and hypertrophy, these effects were confirmed by silencing IRF1 in WT mice, and significantly reversed by overexpression of IRF1 in AIM2<sup>-/-</sup> mice. Taken together, our findings suggest that AIM2 serves as a novel target for the treatment of diabetic cardiomyopathy.</p>\",\"PeriodicalId\":15224,\"journal\":{\"name\":\"Journal of Cardiovascular Translational Research\",\"volume\":\" \",\"pages\":\"94-109\"},\"PeriodicalIF\":2.5000,\"publicationDate\":\"2025-02-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Cardiovascular Translational Research\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1007/s12265-024-10556-0\",\"RegionNum\":3,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/9/4 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"CARDIAC & CARDIOVASCULAR SYSTEMS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Cardiovascular Translational Research","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s12265-024-10556-0","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/9/4 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"CARDIAC & CARDIOVASCULAR SYSTEMS","Score":null,"Total":0}
AIM2 Deficiency Alleviates Cardiac Inflammation and Hypertrophy in HFD/STZ-Induced Diabetic Mice by Inhibiting the NLRC4/IRF1 Signaling Pathway.
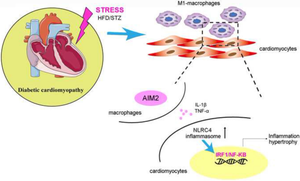
Absent in melanoma 2(AIM2) exacerbates atherosclerosis by inflammasome assembly. However, AIM2-mediated inflammation in diabetic cardiomyopathy remains incompletely understood. Here we investigate the role of AIM2 in high glucose (HG)- and diabetes-induced inflammatory cardiomyopathy. By RNA-seq, we found that AIM2 were significantly upregulated in HG-induced macrophages, upregulation of AIM2 in cardiac infiltrating macrophages was confirmed in a high-fat diet (HFD)/streptozotocin (STZ)-induceddiabetic mouse model . Therefore, AIM2 knockout mice were constructed. Compared to WT mice, HFD/STZ-induced cardiac hypertrophy and dysfunction were significantly improved in AIM2-/- mice, despite no changes in blood glucose and body weight. Further, AIM2 deficiency inhibited cardiac recruitment of M1-macrophages and cytokine production. Mechanistically, AIM2-deficient macrophgaes reduced IL-1β and TNF-α secretion, which impaired the NLRC4/IRF1 signaling in cardiomyocytes, and reduced further recruitment of macrophages, attenuated cardiac inflammation and hypertrophy, these effects were confirmed by silencing IRF1 in WT mice, and significantly reversed by overexpression of IRF1 in AIM2-/- mice. Taken together, our findings suggest that AIM2 serves as a novel target for the treatment of diabetic cardiomyopathy.
期刊介绍:
Journal of Cardiovascular Translational Research (JCTR) is a premier journal in cardiovascular translational research.
JCTR is the journal of choice for authors seeking the broadest audience for emerging technologies, therapies and diagnostics, pre-clinical research, and first-in-man clinical trials.
JCTR''s intent is to provide a forum for critical evaluation of the novel cardiovascular science, to showcase important and clinically relevant aspects of the new research, as well as to discuss the impediments that may need to be overcome during the translation to patient care.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


