Jenni Laitila, Robert A. E. Seaborne, Natasha Ranu, Justin S. Kolb, Carina Wallgren-Pettersson, Nanna Witting, John Vissing, Juan Jesus Vilchez, Edmar Zanoteli, Johanna Palmio, Sanna Huovinen, Henk Granzier, Julien Ochala
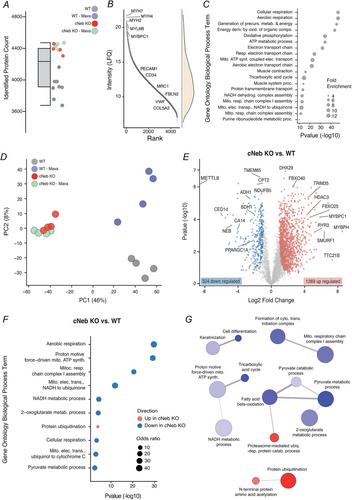
{"title":"肌球蛋白ATP酶抑制未能挽救球蛋白缺陷肌肉代谢失调的蛋白质组。","authors":"Jenni Laitila, Robert A. E. Seaborne, Natasha Ranu, Justin S. Kolb, Carina Wallgren-Pettersson, Nanna Witting, John Vissing, Juan Jesus Vilchez, Edmar Zanoteli, Johanna Palmio, Sanna Huovinen, Henk Granzier, Julien Ochala","doi":"10.1113/JP286870","DOIUrl":null,"url":null,"abstract":"<div>\n \n <section>\n \n \n <div>Nemaline myopathy (NM) is a genetic muscle disease, primarily caused by mutations in the <i>NEB</i> gene (<i>NEB-</i>NM) and with muscle myosin dysfunction as a major molecular pathogenic mechanism. Recently, we have observed that the myosin biochemical super-relaxed state was significantly impaired in <i>NEB</i>-NM, inducing an aberrant increase in ATP consumption and remodelling of the energy proteome in diseased muscle fibres. Because the small-molecule Mavacamten is known to promote the myosin super-relaxed state and reduce the ATP demand, we tested its potency in the context of <i>NEB</i>-NM. We first conducted <i>in vitro</i> experiments in isolated single myofibres from patients and found that Mavacamten successfully reversed the myosin ATP overconsumption. Following this, we assessed its short-term <i>in vivo</i> effects using the conditional nebulin knockout (c<i>Neb</i> KO) mouse model and subsequently performing global proteomics profiling in dissected soleus myofibres. After a 4 week treatment period, we observed a remodelling of a large number of proteins in both c<i>Neb</i> KO mice and their wild-type siblings. Nevertheless, these changes were not related to the energy proteome, indicating that short-term Mavacamten treatment is not sufficient to properly counterbalance the metabolically dysregulated proteome of c<i>Neb</i> KO mice. Taken together, our findings emphasize Mavacamten potency <i>in vitro</i> but challenge its short-term efficacy <i>in vivo</i>.\n\n <figure>\n <div><picture>\n <source></source></picture><p></p>\n </div>\n </figure>\n </div>\n </section>\n \n <section>\n \n <h3> Key points</h3>\n \n <div>\n <ul>\n \n <li>No cure exists for nemaline myopathy, a type of genetic skeletal muscle disease mainly derived from mutations in genes encoding myofilament proteins.</li>\n \n <li>Applying Mavacamten, a small molecule directly targeting the myofilaments, to isolated membrane-permeabilized muscle fibres from human patients restored myosin energetic disturbances.</li>\n \n <li>Treating a mouse model of nemaline myopathy <i>in vivo</i> with Mavacamten for 4 weeks, remodelled the skeletal muscle fibre proteome without any noticeable effects on energetic proteins.</li>\n \n <li>Short-term Mavacamten treatment may not be sufficient to reverse the muscle phenotype in nemaline myopathy.</li>\n </ul>\n </div>\n </section>\n </div>","PeriodicalId":50088,"journal":{"name":"Journal of Physiology-London","volume":"602 20","pages":"5229-5245"},"PeriodicalIF":4.7000,"publicationDate":"2024-08-31","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1113/JP286870","citationCount":"0","resultStr":"{\"title\":\"Myosin ATPase inhibition fails to rescue the metabolically dysregulated proteome of nebulin-deficient muscle\",\"authors\":\"Jenni Laitila, Robert A. E. Seaborne, Natasha Ranu, Justin S. Kolb, Carina Wallgren-Pettersson, Nanna Witting, John Vissing, Juan Jesus Vilchez, Edmar Zanoteli, Johanna Palmio, Sanna Huovinen, Henk Granzier, Julien Ochala\",\"doi\":\"10.1113/JP286870\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div>\\n \\n <section>\\n \\n \\n <div>Nemaline myopathy (NM) is a genetic muscle disease, primarily caused by mutations in the <i>NEB</i> gene (<i>NEB-</i>NM) and with muscle myosin dysfunction as a major molecular pathogenic mechanism. Recently, we have observed that the myosin biochemical super-relaxed state was significantly impaired in <i>NEB</i>-NM, inducing an aberrant increase in ATP consumption and remodelling of the energy proteome in diseased muscle fibres. Because the small-molecule Mavacamten is known to promote the myosin super-relaxed state and reduce the ATP demand, we tested its potency in the context of <i>NEB</i>-NM. We first conducted <i>in vitro</i> experiments in isolated single myofibres from patients and found that Mavacamten successfully reversed the myosin ATP overconsumption. Following this, we assessed its short-term <i>in vivo</i> effects using the conditional nebulin knockout (c<i>Neb</i> KO) mouse model and subsequently performing global proteomics profiling in dissected soleus myofibres. After a 4 week treatment period, we observed a remodelling of a large number of proteins in both c<i>Neb</i> KO mice and their wild-type siblings. Nevertheless, these changes were not related to the energy proteome, indicating that short-term Mavacamten treatment is not sufficient to properly counterbalance the metabolically dysregulated proteome of c<i>Neb</i> KO mice. Taken together, our findings emphasize Mavacamten potency <i>in vitro</i> but challenge its short-term efficacy <i>in vivo</i>.\\n\\n <figure>\\n <div><picture>\\n <source></source></picture><p></p>\\n </div>\\n </figure>\\n </div>\\n </section>\\n \\n <section>\\n \\n <h3> Key points</h3>\\n \\n <div>\\n <ul>\\n \\n <li>No cure exists for nemaline myopathy, a type of genetic skeletal muscle disease mainly derived from mutations in genes encoding myofilament proteins.</li>\\n \\n <li>Applying Mavacamten, a small molecule directly targeting the myofilaments, to isolated membrane-permeabilized muscle fibres from human patients restored myosin energetic disturbances.</li>\\n \\n <li>Treating a mouse model of nemaline myopathy <i>in vivo</i> with Mavacamten for 4 weeks, remodelled the skeletal muscle fibre proteome without any noticeable effects on energetic proteins.</li>\\n \\n <li>Short-term Mavacamten treatment may not be sufficient to reverse the muscle phenotype in nemaline myopathy.</li>\\n </ul>\\n </div>\\n </section>\\n </div>\",\"PeriodicalId\":50088,\"journal\":{\"name\":\"Journal of Physiology-London\",\"volume\":\"602 20\",\"pages\":\"5229-5245\"},\"PeriodicalIF\":4.7000,\"publicationDate\":\"2024-08-31\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1113/JP286870\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Physiology-London\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1113/JP286870\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"NEUROSCIENCES\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Physiology-London","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1113/JP286870","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"NEUROSCIENCES","Score":null,"Total":0}
引用次数: 0
摘要
内膜性肌病(Nemaline myopathy,NM)是一种遗传性肌肉疾病,主要由 NEB 基因突变(NEB-NM)引起,肌肉肌球蛋白功能障碍是其主要的分子致病机制。最近,我们观察到 NEB-NM 中肌球蛋白生化超松弛状态明显受损,诱发 ATP 消耗异常增加和病变肌纤维能量蛋白组重塑。众所周知,小分子 Mavacamten 可促进肌球蛋白超松弛状态并减少 ATP 需求,因此我们测试了其在 NEB-NM 中的有效性。我们首先在分离的患者单个肌纤维中进行了体外实验,发现 Mavacamten 成功逆转了肌球蛋白 ATP 过度消耗。随后,我们使用条件性神经球蛋白基因敲除(cNeb KO)小鼠模型评估了它的短期体内效应,并随后在解剖的比目鱼肌纤维中进行了全局蛋白质组学分析。经过 4 周的治疗后,我们在 cNeb KO 小鼠及其野生型同胞中观察到大量蛋白质发生了重塑。然而,这些变化与能量蛋白质组无关,这表明短期的马伐卡滕治疗不足以适当平衡 cNeb KO 小鼠代谢失调的蛋白质组。综上所述,我们的研究结果强调了 Mavacamten 在体外的有效性,但对其在体内的短期疗效提出了质疑。要点:神经性肌病是一种遗传性骨骼肌疾病,主要由编码肌丝蛋白的基因突变引起,目前尚无治疗方法。将直接靶向肌丝的小分子 Mavacamten 应用于人类患者的分离膜过滤肌纤维,可恢复肌球蛋白的能量紊乱。用 Mavacamten 对神经性肌病小鼠模型进行为期 4 周的体内治疗后,骨骼肌纤维蛋白质组发生了重塑,但对高能蛋白质没有产生任何明显影响。短期 Mavacamten 治疗可能不足以逆转神经性肌病的肌肉表型。
Myosin ATPase inhibition fails to rescue the metabolically dysregulated proteome of nebulin-deficient muscle
Nemaline myopathy (NM) is a genetic muscle disease, primarily caused by mutations in the NEB gene (NEB-NM) and with muscle myosin dysfunction as a major molecular pathogenic mechanism. Recently, we have observed that the myosin biochemical super-relaxed state was significantly impaired in NEB-NM, inducing an aberrant increase in ATP consumption and remodelling of the energy proteome in diseased muscle fibres. Because the small-molecule Mavacamten is known to promote the myosin super-relaxed state and reduce the ATP demand, we tested its potency in the context of NEB-NM. We first conducted in vitro experiments in isolated single myofibres from patients and found that Mavacamten successfully reversed the myosin ATP overconsumption. Following this, we assessed its short-term in vivo effects using the conditional nebulin knockout (cNeb KO) mouse model and subsequently performing global proteomics profiling in dissected soleus myofibres. After a 4 week treatment period, we observed a remodelling of a large number of proteins in both cNeb KO mice and their wild-type siblings. Nevertheless, these changes were not related to the energy proteome, indicating that short-term Mavacamten treatment is not sufficient to properly counterbalance the metabolically dysregulated proteome of cNeb KO mice. Taken together, our findings emphasize Mavacamten potency in vitro but challenge its short-term efficacy in vivo.
Key points
No cure exists for nemaline myopathy, a type of genetic skeletal muscle disease mainly derived from mutations in genes encoding myofilament proteins.
Applying Mavacamten, a small molecule directly targeting the myofilaments, to isolated membrane-permeabilized muscle fibres from human patients restored myosin energetic disturbances.
Treating a mouse model of nemaline myopathy in vivo with Mavacamten for 4 weeks, remodelled the skeletal muscle fibre proteome without any noticeable effects on energetic proteins.
Short-term Mavacamten treatment may not be sufficient to reverse the muscle phenotype in nemaline myopathy.
期刊介绍:
The Journal of Physiology publishes full-length original Research Papers and Techniques for Physiology, which are short papers aimed at disseminating new techniques for physiological research. Articles solicited by the Editorial Board include Perspectives, Symposium Reports and Topical Reviews, which highlight areas of special physiological interest. CrossTalk articles are short editorial-style invited articles framing a debate between experts in the field on controversial topics. Letters to the Editor and Journal Club articles are also published. All categories of papers are subjected to peer reivew.
The Journal of Physiology welcomes submitted research papers in all areas of physiology. Authors should present original work that illustrates new physiological principles or mechanisms. Papers on work at the molecular level, at the level of the cell membrane, single cells, tissues or organs and on systems physiology are all acceptable. Theoretical papers and papers that use computational models to further our understanding of physiological processes will be considered if based on experimentally derived data and if the hypothesis advanced is directly amenable to experimental testing. While emphasis is on human and mammalian physiology, work on lower vertebrate or invertebrate preparations may be suitable if it furthers the understanding of the functioning of other organisms including mammals.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


