{"title":"预测 miRNA 对基因调控的影响,促进多基因转化研究--超级增强子的作用综述。","authors":"Sarmistha Das, Shesh N Rai","doi":"10.3390/ncrna10040045","DOIUrl":null,"url":null,"abstract":"<p><p>Gene regulation is crucial for cellular function and homeostasis. It involves diverse mechanisms controlling the production of specific gene products and contributing to tissue-specific variations in gene expression. The dysregulation of genes leads to disease, emphasizing the need to understand these mechanisms. Computational methods have jointly studied transcription factors (TFs), microRNA (miRNA), and messenger RNA (mRNA) to investigate gene regulatory networks. However, there remains a knowledge gap in comprehending gene regulatory networks. On the other hand, super-enhancers (SEs) have been implicated in miRNA biogenesis and function in recent experimental studies, in addition to their pivotal roles in cell identity and disease progression. However, statistical/computational methodologies harnessing the potential of SEs in deciphering gene regulation networks remain notably absent. However, to understand the effect of miRNA on mRNA, existing statistical/computational methods could be updated, or novel methods could be developed by accounting for SEs in the model. In this review, we categorize existing computational methods that utilize TF and miRNA data to understand gene regulatory networks into three broad areas and explore the challenges of integrating enhancers/SEs. The three areas include unraveling indirect regulatory networks, identifying network motifs, and enriching pathway identification by dissecting gene regulators. We hypothesize that addressing these challenges will enhance our understanding of gene regulation, aiding in the identification of therapeutic targets and disease biomarkers. We believe that constructing statistical/computational models that dissect the role of SEs in predicting the effect of miRNA on gene regulation is crucial for tackling these challenges.</p>","PeriodicalId":19271,"journal":{"name":"Non-Coding RNA","volume":"10 4","pages":""},"PeriodicalIF":3.6000,"publicationDate":"2024-08-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11357235/pdf/","citationCount":"0","resultStr":"{\"title\":\"Predicting the Effect of miRNA on Gene Regulation to Foster Translational Multi-Omics Research-A Review on the Role of Super-Enhancers.\",\"authors\":\"Sarmistha Das, Shesh N Rai\",\"doi\":\"10.3390/ncrna10040045\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Gene regulation is crucial for cellular function and homeostasis. It involves diverse mechanisms controlling the production of specific gene products and contributing to tissue-specific variations in gene expression. The dysregulation of genes leads to disease, emphasizing the need to understand these mechanisms. Computational methods have jointly studied transcription factors (TFs), microRNA (miRNA), and messenger RNA (mRNA) to investigate gene regulatory networks. However, there remains a knowledge gap in comprehending gene regulatory networks. On the other hand, super-enhancers (SEs) have been implicated in miRNA biogenesis and function in recent experimental studies, in addition to their pivotal roles in cell identity and disease progression. However, statistical/computational methodologies harnessing the potential of SEs in deciphering gene regulation networks remain notably absent. However, to understand the effect of miRNA on mRNA, existing statistical/computational methods could be updated, or novel methods could be developed by accounting for SEs in the model. In this review, we categorize existing computational methods that utilize TF and miRNA data to understand gene regulatory networks into three broad areas and explore the challenges of integrating enhancers/SEs. The three areas include unraveling indirect regulatory networks, identifying network motifs, and enriching pathway identification by dissecting gene regulators. We hypothesize that addressing these challenges will enhance our understanding of gene regulation, aiding in the identification of therapeutic targets and disease biomarkers. We believe that constructing statistical/computational models that dissect the role of SEs in predicting the effect of miRNA on gene regulation is crucial for tackling these challenges.</p>\",\"PeriodicalId\":19271,\"journal\":{\"name\":\"Non-Coding RNA\",\"volume\":\"10 4\",\"pages\":\"\"},\"PeriodicalIF\":3.6000,\"publicationDate\":\"2024-08-15\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11357235/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Non-Coding RNA\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.3390/ncrna10040045\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Non-Coding RNA","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3390/ncrna10040045","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
摘要
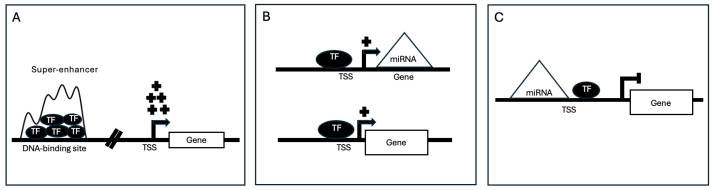
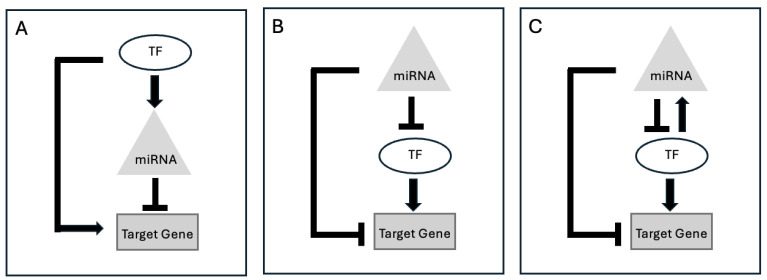
基因调控对细胞功能和平衡至关重要。它涉及多种机制,控制特定基因产物的产生,并导致基因表达的组织特异性变化。基因失调会导致疾病,因此有必要了解这些机制。计算方法联合研究了转录因子(TFs)、微RNA(miRNA)和信使RNA(mRNA),以研究基因调控网络。然而,在理解基因调控网络方面仍存在知识空白。另一方面,在最近的实验研究中,超级增强子(SEs)除了在细胞特性和疾病进展中发挥关键作用外,还与 miRNA 的生物发生和功能有关。然而,利用 SEs 的潜力来破译基因调控网络的统计/计算方法仍然明显缺乏。然而,要了解 miRNA 对 mRNA 的影响,可以更新现有的统计/计算方法,或者通过在模型中考虑 SEs 来开发新的方法。在这篇综述中,我们将利用 TF 和 miRNA 数据了解基因调控网络的现有计算方法分为三大领域,并探讨了整合增强子/SEs 所面临的挑战。这三个领域包括揭示间接调控网络、识别网络母题以及通过剖析基因调控因子丰富通路识别。我们假设,应对这些挑战将加深我们对基因调控的理解,有助于确定治疗靶点和疾病生物标志物。我们相信,构建统计/计算模型,剖析 SE 在预测 miRNA 对基因调控影响方面的作用,对于应对这些挑战至关重要。
Predicting the Effect of miRNA on Gene Regulation to Foster Translational Multi-Omics Research-A Review on the Role of Super-Enhancers.
Gene regulation is crucial for cellular function and homeostasis. It involves diverse mechanisms controlling the production of specific gene products and contributing to tissue-specific variations in gene expression. The dysregulation of genes leads to disease, emphasizing the need to understand these mechanisms. Computational methods have jointly studied transcription factors (TFs), microRNA (miRNA), and messenger RNA (mRNA) to investigate gene regulatory networks. However, there remains a knowledge gap in comprehending gene regulatory networks. On the other hand, super-enhancers (SEs) have been implicated in miRNA biogenesis and function in recent experimental studies, in addition to their pivotal roles in cell identity and disease progression. However, statistical/computational methodologies harnessing the potential of SEs in deciphering gene regulation networks remain notably absent. However, to understand the effect of miRNA on mRNA, existing statistical/computational methods could be updated, or novel methods could be developed by accounting for SEs in the model. In this review, we categorize existing computational methods that utilize TF and miRNA data to understand gene regulatory networks into three broad areas and explore the challenges of integrating enhancers/SEs. The three areas include unraveling indirect regulatory networks, identifying network motifs, and enriching pathway identification by dissecting gene regulators. We hypothesize that addressing these challenges will enhance our understanding of gene regulation, aiding in the identification of therapeutic targets and disease biomarkers. We believe that constructing statistical/computational models that dissect the role of SEs in predicting the effect of miRNA on gene regulation is crucial for tackling these challenges.
Non-Coding RNABiochemistry, Genetics and Molecular Biology-Genetics
CiteScore
6.70
自引率
4.70%
发文量
74
审稿时长
10 weeks
期刊介绍:
Functional studies dealing with identification, structure-function relationships or biological activity of: small regulatory RNAs (miRNAs, siRNAs and piRNAs) associated with the RNA interference pathway small nuclear RNAs, small nucleolar and tRNAs derived small RNAs other types of small RNAs, such as those associated with splice junctions and transcription start sites long non-coding RNAs, including antisense RNAs, long ''intergenic'' RNAs, intronic RNAs and ''enhancer'' RNAs other classes of RNAs such as vault RNAs, scaRNAs, circular RNAs, 7SL RNAs, telomeric and centromeric RNAs regulatory functions of mRNAs and UTR-derived RNAs catalytic and allosteric (riboswitch) RNAs viral, transposon and repeat-derived RNAs bacterial regulatory RNAs, including CRISPR RNAS Analysis of RNA processing, RNA binding proteins, RNA signaling and RNA interaction pathways: DICER AGO, PIWI and PIWI-like proteins other classes of RNA binding and RNA transport proteins RNA interactions with chromatin-modifying complexes RNA interactions with DNA and other RNAs the role of RNA in the formation and function of specialized subnuclear organelles and other aspects of cell biology intercellular and intergenerational RNA signaling RNA processing structure-function relationships in RNA complexes RNA analyses, informatics, tools and technologies: transcriptomic analyses and technologies development of tools and technologies for RNA biology and therapeutics Translational studies involving long and short non-coding RNAs: identification of biomarkers development of new therapies involving microRNAs and other ncRNAs clinical studies involving microRNAs and other ncRNAs.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


