{"title":"深度学习势能辅助预测 SrCl2-KCl-MgCl2 熔体的局部结构和热物理性质。","authors":"Jia Zhao, Taixi Feng, Guimin Lu","doi":"10.1021/acs.jctc.4c00824","DOIUrl":null,"url":null,"abstract":"<p><p>The local structure and thermophysical properties of SrCl<sub>2</sub>-KCl-MgCl<sub>2</sub> melt were revealed by deep potential molecular dynamicsdriven by machine learning to facilitate the development of molten salt electrolytic Mg-Sr alloys. The short- and intermediate-range order of the SrCl<sub>2</sub>-KCl-MgCl<sub>2</sub> melts was explored through radial distribution functions and structure factors, respectively, and their component and temperature dependence were discussed comprehensively. In the MgCl<sub>2</sub>-rich system, the intermediate-range order is more pronounced, and its evolution with temperature exhibits a non-Debye-Waller behavior. Mg-Cl is dominated by 4,5 coordination and Sr-Cl by 6,7 coordination, and their coordination geometries exhibit distorted octahedra and distorted pentagonal bipyramids, respectively. A database of thermophysical properties of SrCl<sub>2</sub>-KCl-MgCl<sub>2</sub> melts, including density, self-diffusion coefficient, viscosity, and ionic conductivity, was thus developed, covering the temperature range from 873 to 1173 K.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":" ","pages":"7611-7623"},"PeriodicalIF":5.7000,"publicationDate":"2024-09-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Deep Learning Potential Assisted Prediction of Local Structure and Thermophysical Properties of the SrCl<sub>2</sub>-KCl-MgCl<sub>2</sub> Melt.\",\"authors\":\"Jia Zhao, Taixi Feng, Guimin Lu\",\"doi\":\"10.1021/acs.jctc.4c00824\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>The local structure and thermophysical properties of SrCl<sub>2</sub>-KCl-MgCl<sub>2</sub> melt were revealed by deep potential molecular dynamicsdriven by machine learning to facilitate the development of molten salt electrolytic Mg-Sr alloys. The short- and intermediate-range order of the SrCl<sub>2</sub>-KCl-MgCl<sub>2</sub> melts was explored through radial distribution functions and structure factors, respectively, and their component and temperature dependence were discussed comprehensively. In the MgCl<sub>2</sub>-rich system, the intermediate-range order is more pronounced, and its evolution with temperature exhibits a non-Debye-Waller behavior. Mg-Cl is dominated by 4,5 coordination and Sr-Cl by 6,7 coordination, and their coordination geometries exhibit distorted octahedra and distorted pentagonal bipyramids, respectively. A database of thermophysical properties of SrCl<sub>2</sub>-KCl-MgCl<sub>2</sub> melts, including density, self-diffusion coefficient, viscosity, and ionic conductivity, was thus developed, covering the temperature range from 873 to 1173 K.</p>\",\"PeriodicalId\":45,\"journal\":{\"name\":\"Journal of Chemical Theory and Computation\",\"volume\":\" \",\"pages\":\"7611-7623\"},\"PeriodicalIF\":5.7000,\"publicationDate\":\"2024-09-10\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Theory and Computation\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://doi.org/10.1021/acs.jctc.4c00824\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/8/28 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jctc.4c00824","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/8/28 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Deep Learning Potential Assisted Prediction of Local Structure and Thermophysical Properties of the SrCl2-KCl-MgCl2 Melt.
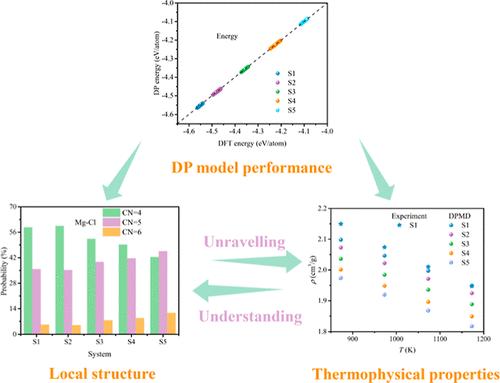
The local structure and thermophysical properties of SrCl2-KCl-MgCl2 melt were revealed by deep potential molecular dynamicsdriven by machine learning to facilitate the development of molten salt electrolytic Mg-Sr alloys. The short- and intermediate-range order of the SrCl2-KCl-MgCl2 melts was explored through radial distribution functions and structure factors, respectively, and their component and temperature dependence were discussed comprehensively. In the MgCl2-rich system, the intermediate-range order is more pronounced, and its evolution with temperature exhibits a non-Debye-Waller behavior. Mg-Cl is dominated by 4,5 coordination and Sr-Cl by 6,7 coordination, and their coordination geometries exhibit distorted octahedra and distorted pentagonal bipyramids, respectively. A database of thermophysical properties of SrCl2-KCl-MgCl2 melts, including density, self-diffusion coefficient, viscosity, and ionic conductivity, was thus developed, covering the temperature range from 873 to 1173 K.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


