基于甲基化相关 lncRNA 的肺腺癌预测模型
摘要
背景:甲基化与肺腺癌(LUAD)的发生和发展之间的关系已接近尾声。长非编码 RNA(lncRNA)作为多种生物功能的调控因子,可用于癌症诊断。我们的研究旨在构建与甲基化相关的LUAD lncRNA特征:在癌症基因组图谱(TCGA)数据集中,我们下载了LUAD病例的RNA表达数据和临床信息。为了建立基于甲基化相关lncRNA的最佳预后特征,我们采用了Cox回归分析。通过Kaplan-Meier分析,比较了不同风险类别(包括低风险和高风险患者)的总生存率。为了根据基因的功能意义对其进行分类,采用了 GSEA(Subramanian 等人,2005 年)。单样本基因组富集分析(ssGSEA)被用来进一步揭示甲基化相关lncRNA预后模型在免疫浸润中的潜在分子机制。利用TRLnc(http://www.licpathway.net/TRlnc)和lncRNASNP分析这18个lncRNA的SNP位点和TRLnc。利用 LncSEA 网站分析肿瘤发生和发展过程中的 18 个 lncRNA。Go 用于分析这 18 个 lncRNA 的 TF(转录因子)、Cerna 网络和相互结合的蛋白质所富集的通路。预言 "软件包用于分析这一预后模型在指导个性化免疫疗法方面的价值:在这项研究中,我们发现了18个与甲基化相关的lncRNA(AP002761.1、AL118558.3、CH17-340M24.3、AL353150.1、AC004687.1、LINC00996、AF186192.1、HSPC324、AC087752.3、FAM30A、AC106047.1、AC026355.1、ABALON、LINC01843、AL606489.1、NKILA、AP001453.2、GSEC)来建立一个甲基化相关的lncRNA特征,以检测LUAD患者的预后。与18个lncRNA相互作用的蛋白质所富集的通路主要是EMT、缺氧、干性和增殖,其中LINC00996和AF186192.1受TP53和TP63等多种肿瘤相关转录因子调控,fam30a与mRNA形成Cerna网络。LINC00996中有2319个SNP位点,其中36个为风险SNP位点,af186192.1中有205个SNP位点;AF186192.1影响95个保守miRNA和123个非保守miRNA,促进149对miRNA:lncRNA的结合,抑制95对miRNA:lncRNA的结合。ROC曲线显示,已建立的甲基化相关lncRNA特征在预测LUAD患者预后方面比临床病理参数更有效。我们的研究证实,基于甲基化相关lncRNA的风险评分模型分出的高危组患者的OS较短。根据GSEA,高危组主要富集了肿瘤和免疫相关通路。ssGSEA显示,LUAD患者的预测特征与免疫状态之间存在明显关联。此外,主成分分析(PCA)证明了我们的特征具有预后和预测价值。甲基化相关lncRNA预测特征与常规化疗药物IC50之间的相关性可为LUAD患者提供个性化的化疗方案。甲基化相关lncRNA特征能有效预测LUAD患者的DFS。
Background
The collaboration between methylation and the lung adenocarcinoma (LUAD) occurrence and development is closes. Long noncoding RNA (lncRNA), as a regulatory factor of various biological functions, can be used for cancer diagnosis. Our study aimed to construct a robust methylation-related lncRNA signature of LUAD.
Methods
In the Cancer Genome Atlas (TCGA) dataset, we download the RNA expression data and clinical information of LUAD cases. To develop the best prognostic signature based on methylation-related lncRNAs, Cox regression analyses were utilized. Using Kaplan–Meier analysis, overall survival rates were compared between risk category included both low- and high-risk patients. To categorize genes according to their functional significance, GSEA (Subramanian et al, 2005) was used. Single-sample gene set enrichment analysis (ssGSEA) was used to further reveal the potential molecular mechanism of the methylation-related lncRNA prognostic model in immune infiltration. Using TRLnc (http://www.licpathway.net/TRlnc) and lncRNASNP to analyse the SNP sites and TRLnc of these 18 lncRNAs. LncSEA website was used to analyse 18 lncRNA in the process of tumour development and development. Go was used to analyse the enriched pathways enriched by TFs (transcription factors), Cerna networks, and proteins bound to each other of these 18 lncRNAs. The ‘prophetic’ package was used to analyse the value of this prognostic model in guiding personalized immunotherapy.
Results
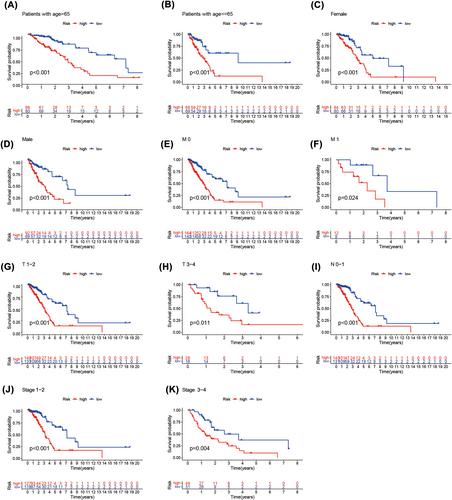
In this study, we identified 18 methylation-related lncRNAs (AP002761.1, AL118558.3, CH17-340M24.3, AL353150.1, AC004687.1, LINC00996, AF186192.1, HSPC324, AC087752.3, FAM30A, AC106047.1, AC026355.1, ABALON, LINC01843, AL606489.1, NKILA, AP001453.2, GSEC) to establish a methylation-related lncRNA signature that can detect patients prognosis in LUAD. The enriched pathways enriched by proteins interacting with 18 lncRNAs are mainly EMT, hypoxia, stemness and proliferation, among which LINC00996 and AF186192.1 are regulated by multiple tumour associated transcription factors, such as TP53 and TP63, and fam30a and mRNA form a Cerna network. There are 2319 SNP loci in LINC00996, 36 of which are risk SNP loci and 205 SNP loci in af186192.1; AF186192.1 affects 95 conserved miRNAs and 123 non-conserved miRNAs, promotes the binding of 149 pairs of miRNAs: lncRNAs and inhibits the binding of 95 pairs of miRNAs: lncRNAs. The ROC curve demonstrated that the established methylation-related lncRNA signature was more effective in predicting the prognosis of patients in LUAD than the clinicopathological parameters. Our research has confirmed that patients in the high-risk group which was separated by the risk score model based on methylation-related lncRNA had shorter OS. According to GSEA, the high-risk group had a predominantly tumour- and immune-related pathway enrichment. A significant association was shown by ssGSEA between predictive signature and immune status in LUAD patients. In addition, principal component analysis (PCA) demonstrated the prognostic and predictive value of our signature. The correlation between the predictive signature of methylation-related lncRNA and IC50 of conventional chemotherapy drugs can provide personalized chemotherapy regimens for LUAD patients. Methylation-related lncRNA signature can effectively predict DFS of patients in LUAD.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


