{"title":"关于钴在噻吩吸附小钼和钼钴簇上的作用的计算研究,作为加氢脱硫过程的位点模型","authors":"Julián Del Plá, Reinaldo Pis Diez","doi":"10.1016/j.susc.2024.122571","DOIUrl":null,"url":null,"abstract":"<div><p>Non periodic density functional theory calculations are used to investigate the role of cobalt atoms in the adsorption of thiophene on small Mo and MoCo clusters. Metallic aggregates play the role of those active sites found in the true catalysts. Two interaction modes between thiophene and metallic sites are considered, namely, the S-mode, in which the organosulfur molecule interacts through the S atom, and the R-mode, in which the interaction takes place through the thiophene ring. A large number of sites, in which thiophene effectively adsorbs, was found, both in the monometallic case and in the bimetallic one. Considerably larger adsorption energies were found when thiophene interacts via the R-mode than when adsorption occurs through the S-mode. The activation of C-S bonds is also more important for R-mode cases than for S-mode ones. Further analysis made on some selected systems and based on density of states and molecular orbital overlap population-projected density of states reveals that thiophene and metallic clusters interact in an energy range around −6.0 eV with respect to the Fermi energy. Bands observed at energies below −6.0 eV correspond to thiophene states that become shifted with respect to the values obtained for isolated thiophene depending on the strength of the interaction. Bands above -6.0 eV describe how C and S atoms interact with Co and Mo ones, providing both bonding and antibonding patterns that helps to understand the overall interaction. Most important is the finding that cobalt atoms seem to play no relevant role during the adsorption of thiophene on metallic sites. Thus, present results obtained using non periodic GGA density functional theory seem to point to cobalt taking part in another step of the overall HDS process, hydrogen adsorption or hydrogen attack to C-S bonds, for instance.</p></div>","PeriodicalId":22100,"journal":{"name":"Surface Science","volume":"749 ","pages":"Article 122571"},"PeriodicalIF":1.8000,"publicationDate":"2024-08-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"A computational study of the role of cobalt in thiophene adsorption on small Mo and MoCo clusters as site models for the HDS process\",\"authors\":\"Julián Del Plá, Reinaldo Pis Diez\",\"doi\":\"10.1016/j.susc.2024.122571\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>Non periodic density functional theory calculations are used to investigate the role of cobalt atoms in the adsorption of thiophene on small Mo and MoCo clusters. Metallic aggregates play the role of those active sites found in the true catalysts. Two interaction modes between thiophene and metallic sites are considered, namely, the S-mode, in which the organosulfur molecule interacts through the S atom, and the R-mode, in which the interaction takes place through the thiophene ring. A large number of sites, in which thiophene effectively adsorbs, was found, both in the monometallic case and in the bimetallic one. Considerably larger adsorption energies were found when thiophene interacts via the R-mode than when adsorption occurs through the S-mode. The activation of C-S bonds is also more important for R-mode cases than for S-mode ones. Further analysis made on some selected systems and based on density of states and molecular orbital overlap population-projected density of states reveals that thiophene and metallic clusters interact in an energy range around −6.0 eV with respect to the Fermi energy. Bands observed at energies below −6.0 eV correspond to thiophene states that become shifted with respect to the values obtained for isolated thiophene depending on the strength of the interaction. Bands above -6.0 eV describe how C and S atoms interact with Co and Mo ones, providing both bonding and antibonding patterns that helps to understand the overall interaction. Most important is the finding that cobalt atoms seem to play no relevant role during the adsorption of thiophene on metallic sites. Thus, present results obtained using non periodic GGA density functional theory seem to point to cobalt taking part in another step of the overall HDS process, hydrogen adsorption or hydrogen attack to C-S bonds, for instance.</p></div>\",\"PeriodicalId\":22100,\"journal\":{\"name\":\"Surface Science\",\"volume\":\"749 \",\"pages\":\"Article 122571\"},\"PeriodicalIF\":1.8000,\"publicationDate\":\"2024-08-14\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Surface Science\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S0039602824001225\",\"RegionNum\":4,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Surface Science","FirstCategoryId":"92","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0039602824001225","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
摘要
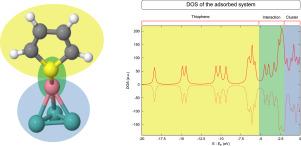
非周期性密度泛函理论计算用于研究钴原子在小钼和钼钴团簇吸附噻吩过程中的作用。金属团聚体扮演了真正催化剂中活性位点的角色。研究考虑了噻吩与金属位点之间的两种相互作用模式,即有机硫分子通过 S 原子相互作用的 S 模式和通过噻吩环相互作用的 R 模式。在单金属和双金属情况下,都发现了大量噻吩有效吸附的位点。当噻吩通过 R 模式相互作用时,吸附能明显大于通过 S 模式吸附时。C-S 键的活化在 R 模式情况下也比在 S 模式情况下更重要。根据状态密度和分子轨道重叠群体推算的状态密度对一些选定的系统进行的进一步分析表明,噻吩和金属团簇在费米能-6.0 eV 左右的能量范围内相互作用。在能量低于 -6.0 eV 时观察到的条带对应于噻吩态,这些噻吩态相对于孤立噻吩态的值会发生偏移,这取决于相互作用的强度。高于 -6.0 eV 的带描述了 C 原子和 S 原子如何与 Co 原子和 Mo 原子相互作用,提供了成键和反键模式,有助于理解整体相互作用。最重要的发现是,钴原子在金属位点吸附噻吩的过程中似乎没有发挥相关作用。因此,利用非周期性 GGA 密度泛函理论获得的当前结果似乎表明,钴参与了整个加氢脱硫过程的另一个步骤,例如氢吸附或氢对 C-S 键的攻击。
A computational study of the role of cobalt in thiophene adsorption on small Mo and MoCo clusters as site models for the HDS process
Non periodic density functional theory calculations are used to investigate the role of cobalt atoms in the adsorption of thiophene on small Mo and MoCo clusters. Metallic aggregates play the role of those active sites found in the true catalysts. Two interaction modes between thiophene and metallic sites are considered, namely, the S-mode, in which the organosulfur molecule interacts through the S atom, and the R-mode, in which the interaction takes place through the thiophene ring. A large number of sites, in which thiophene effectively adsorbs, was found, both in the monometallic case and in the bimetallic one. Considerably larger adsorption energies were found when thiophene interacts via the R-mode than when adsorption occurs through the S-mode. The activation of C-S bonds is also more important for R-mode cases than for S-mode ones. Further analysis made on some selected systems and based on density of states and molecular orbital overlap population-projected density of states reveals that thiophene and metallic clusters interact in an energy range around −6.0 eV with respect to the Fermi energy. Bands observed at energies below −6.0 eV correspond to thiophene states that become shifted with respect to the values obtained for isolated thiophene depending on the strength of the interaction. Bands above -6.0 eV describe how C and S atoms interact with Co and Mo ones, providing both bonding and antibonding patterns that helps to understand the overall interaction. Most important is the finding that cobalt atoms seem to play no relevant role during the adsorption of thiophene on metallic sites. Thus, present results obtained using non periodic GGA density functional theory seem to point to cobalt taking part in another step of the overall HDS process, hydrogen adsorption or hydrogen attack to C-S bonds, for instance.
期刊介绍:
Surface Science is devoted to elucidating the fundamental aspects of chemistry and physics occurring at a wide range of surfaces and interfaces and to disseminating this knowledge fast. The journal welcomes a broad spectrum of topics, including but not limited to:
• model systems (e.g. in Ultra High Vacuum) under well-controlled reactive conditions
• nanoscale science and engineering, including manipulation of matter at the atomic/molecular scale and assembly phenomena
• reactivity of surfaces as related to various applied areas including heterogeneous catalysis, chemistry at electrified interfaces, and semiconductors functionalization
• phenomena at interfaces relevant to energy storage and conversion, and fuels production and utilization
• surface reactivity for environmental protection and pollution remediation
• interactions at surfaces of soft matter, including polymers and biomaterials.
Both experimental and theoretical work, including modeling, is within the scope of the journal. Work published in Surface Science reaches a wide readership, from chemistry and physics to biology and materials science and engineering, providing an excellent forum for cross-fertilization of ideas and broad dissemination of scientific discoveries.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


