{"title":"7p22.3 微缺失:先天性心脏病、神经发育迟缓和癫痫患者的病例研究。","authors":"Liliya Skvortsova, Anastassiya Perfilyeva, Kira Bespalova, Yelena Kuzovleva, Nailya Kabysheva, Ozada Khamdiyeva","doi":"10.1186/s13023-024-03321-8","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Chromosome 7 has regions enriched with low copy repeats (LCRs), which increase the likelihood of chromosomal microdeletion disorders. Documented microdeletion disorders on chromosome 7 include both well-known Williams syndrome and more rare cases. It is noteworthy that most cases of various microdeletions are characterized by phenotypic signs of neuropsychological developmental disorders, which, however, have a different genetic origin. The localization of the microdeletions, the genes included in the region, as well as the structural features of the sequences of these genes have a cumulative influence on the phenotypic characteristics of the individuals for each specific case and the severity of the manifestations of disorders. The consideration of these features and their detailed analysis is important for a correct and comprehensive assessment of the disease.</p><p><strong>Results: </strong>The article describes a clinical case of 7p22.3 microdeletion in a patient with congenital heart defect and neurological abnormalities - epilepsy, combined with moderate mental and motor developmental delay.</p><p><strong>Conclusions: </strong>Through detailed genetic analyses, we are improving the clinical description of the rare 7p22.3 microdeletion and thus creating a basis for future genetic counseling and research into targeted therapies.</p>","PeriodicalId":19651,"journal":{"name":"Orphanet Journal of Rare Diseases","volume":"19 1","pages":"301"},"PeriodicalIF":3.5000,"publicationDate":"2024-08-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11330011/pdf/","citationCount":"0","resultStr":"{\"title\":\"7p22.3 microdeletion: a case study of a patient with congenital heart defect, neurodevelopmental delay and epilepsy.\",\"authors\":\"Liliya Skvortsova, Anastassiya Perfilyeva, Kira Bespalova, Yelena Kuzovleva, Nailya Kabysheva, Ozada Khamdiyeva\",\"doi\":\"10.1186/s13023-024-03321-8\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Chromosome 7 has regions enriched with low copy repeats (LCRs), which increase the likelihood of chromosomal microdeletion disorders. Documented microdeletion disorders on chromosome 7 include both well-known Williams syndrome and more rare cases. It is noteworthy that most cases of various microdeletions are characterized by phenotypic signs of neuropsychological developmental disorders, which, however, have a different genetic origin. The localization of the microdeletions, the genes included in the region, as well as the structural features of the sequences of these genes have a cumulative influence on the phenotypic characteristics of the individuals for each specific case and the severity of the manifestations of disorders. The consideration of these features and their detailed analysis is important for a correct and comprehensive assessment of the disease.</p><p><strong>Results: </strong>The article describes a clinical case of 7p22.3 microdeletion in a patient with congenital heart defect and neurological abnormalities - epilepsy, combined with moderate mental and motor developmental delay.</p><p><strong>Conclusions: </strong>Through detailed genetic analyses, we are improving the clinical description of the rare 7p22.3 microdeletion and thus creating a basis for future genetic counseling and research into targeted therapies.</p>\",\"PeriodicalId\":19651,\"journal\":{\"name\":\"Orphanet Journal of Rare Diseases\",\"volume\":\"19 1\",\"pages\":\"301\"},\"PeriodicalIF\":3.5000,\"publicationDate\":\"2024-08-16\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11330011/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Orphanet Journal of Rare Diseases\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1186/s13023-024-03321-8\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Orphanet Journal of Rare Diseases","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s13023-024-03321-8","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
7p22.3 microdeletion: a case study of a patient with congenital heart defect, neurodevelopmental delay and epilepsy.
Background: Chromosome 7 has regions enriched with low copy repeats (LCRs), which increase the likelihood of chromosomal microdeletion disorders. Documented microdeletion disorders on chromosome 7 include both well-known Williams syndrome and more rare cases. It is noteworthy that most cases of various microdeletions are characterized by phenotypic signs of neuropsychological developmental disorders, which, however, have a different genetic origin. The localization of the microdeletions, the genes included in the region, as well as the structural features of the sequences of these genes have a cumulative influence on the phenotypic characteristics of the individuals for each specific case and the severity of the manifestations of disorders. The consideration of these features and their detailed analysis is important for a correct and comprehensive assessment of the disease.
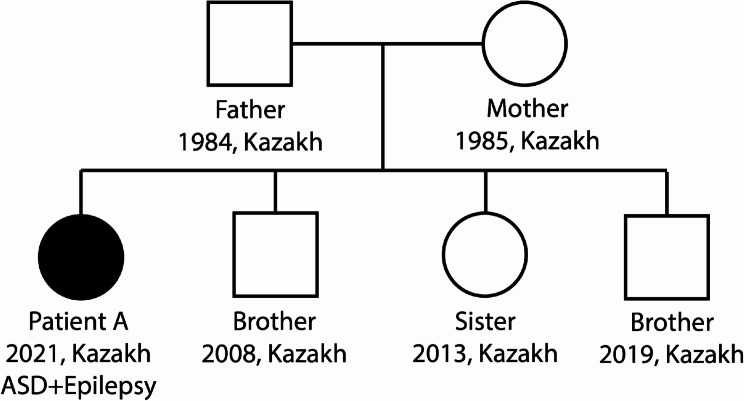
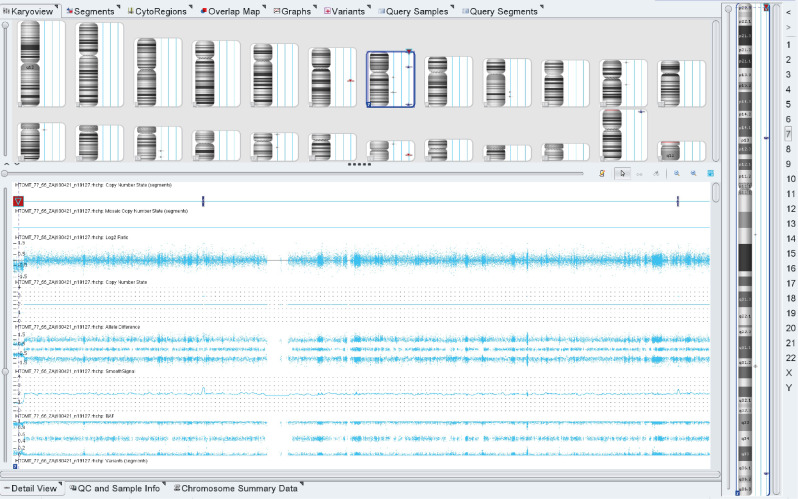
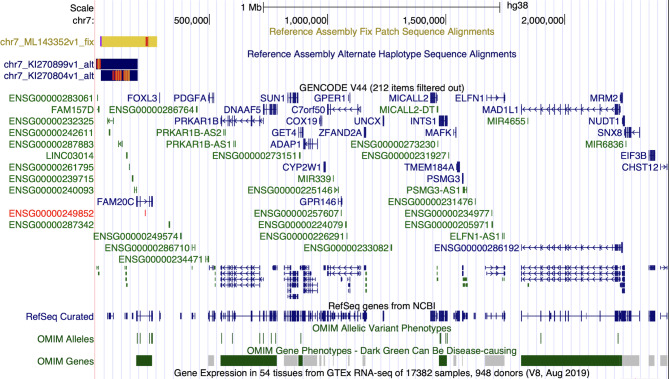
Results: The article describes a clinical case of 7p22.3 microdeletion in a patient with congenital heart defect and neurological abnormalities - epilepsy, combined with moderate mental and motor developmental delay.
Conclusions: Through detailed genetic analyses, we are improving the clinical description of the rare 7p22.3 microdeletion and thus creating a basis for future genetic counseling and research into targeted therapies.
期刊介绍:
Orphanet Journal of Rare Diseases is an open access, peer-reviewed journal that encompasses all aspects of rare diseases and orphan drugs. The journal publishes high-quality reviews on specific rare diseases. In addition, the journal may consider articles on clinical trial outcome reports, either positive or negative, and articles on public health issues in the field of rare diseases and orphan drugs. The journal does not accept case reports.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


