日本和韩国的原发性睫状肌运动障碍是由具有 3000 年历史的 DRC1 基因创始人变异引起的。
IF 2.6
3区 生物学
Q2 GENETICS & HEREDITY
引用次数: 0
摘要
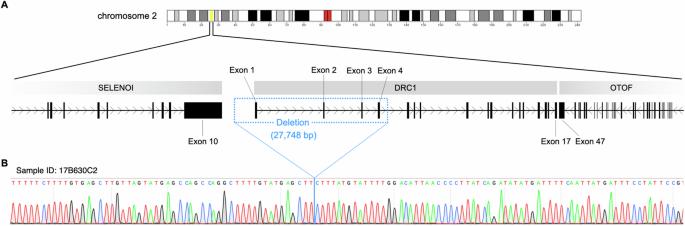
原发性睫状肌运动障碍(PCD)是一种以睫状肌结构异常和功能障碍为特征的遗传性疾病,可导致慢性鼻炎、中耳炎伴渗出液、支气管扩张和不育症。在日本的 PCD 病例中,约有一半归因于动力蛋白调节复合体亚基 1(DRC1)基因的变异,主要表现为第 1-4 号外显子的同质缺失,跨度为 27,748 碱基对,位于第 2 号染色体上。在此,我们报告了 10 例新的 PCD 病例(9 个家庭),以及之前报告的 29 例由 DRC1 基因变异引起的病例(24 个家庭)。在这 39 个病例中,有 38 个病例(97.4%)检测到 DRC1 第 1-4 号外显子双重复缺失。在日本和韩国人群中的所有 PCD 病例中,这些 DRC1 基因缺失都表现出相同的断点,这有力地表明这是一种奠基效应。在本研究中,我们利用全外显子组测序数据集对 18 例携带大型双侧 DRC1 缺失的日本 PCD 患者进行了单倍型分析。我们估计创始等位基因可能出现在 115.1 代以前(95% 置信区间:33.7-205.1),这表明其起源于大约 3050 年前,与日本从绳文时代向弥生时代早期的过渡相吻合。考虑到现代日本人口的形成,带有 DRC1 外显子 1-4 缺失的始祖很可能生活在朝鲜半岛,后来通过移民将等位基因传到了日本。这项研究深入揭示了日本和韩国人群中最常见的 PCD 变异--DRC1 拷贝数变异的起源,强调了在人类迁徙和疾病流行的背景下了解人群特异性遗传变异的重要性。本文章由计算机程序翻译,如有差异,请以英文原文为准。

A 3000-year-old founder variant in the DRC1 gene causes primary ciliary dyskinesia in Japan and Korea
Primary ciliary dyskinesia (PCD) is a genetic disorder characterized by ciliary structural abnormalities and dysfunction, leading to chronic rhinosinusitis, otitis media with effusion, bronchiectasis, and infertility. Approximately half of Japanese PCD cases are attributed to variants in the dynein regulatory complex subunit 1 (DRC1) gene, predominantly featuring homogeneous deletions of exons 1–4 spanning 27,748 base pairs on chromosome 2. Here, we report 10 new PCD cases (9 families) in addition to 29 previously reported cases (24 families) caused by DRC1 variants. Among these 39 cases, biallelic DRC1 exon 1–4 deletions were detected in 38 (97.4%). These DRC1 deletions exhibited an identical breakpoint in all PCD cases in the Japanese and Korean populations, strongly suggesting a founder effect. In this study, we performed haplotype analysis, using a whole-exome sequencing dataset of 18 Japanese PCD patients harboring large biallelic DRC1 deletions. We estimated that the founder allele likely emerged 115.1 generations ago (95% confidence interval: 33.7–205.1), suggesting an origin of approximately 3050 years ago, coinciding with the transition from the Jomon period to the early Yayoi period in Japan. Considering the formation of the modern Japanese population, the founder with the DRC1 exon 1–4 deletion likely lived on the Korean peninsula, with the allele later transmitted to Japan through migration. This study provides insights into the origin of the DRC1 copy number variant, the most frequent PCD variant in the Japanese and Korean populations, highlighting the importance of understanding population-specific genetic variations in the context of human migration and disease prevalence.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Journal of Human Genetics
生物-遗传学
CiteScore
7.20
自引率
0.00%
发文量
101
审稿时长
4-8 weeks
期刊介绍:
The Journal of Human Genetics is an international journal publishing articles on human genetics, including medical genetics and human genome analysis. It covers all aspects of human genetics, including molecular genetics, clinical genetics, behavioral genetics, immunogenetics, pharmacogenomics, population genetics, functional genomics, epigenetics, genetic counseling and gene therapy.
Articles on the following areas are especially welcome: genetic factors of monogenic and complex disorders, genome-wide association studies, genetic epidemiology, cancer genetics, personal genomics, genotype-phenotype relationships and genome diversity.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


