Stefanie Perrier, Julia Macintosh, Agata D. Misiaszek, Gabrielle Lambert, Kether Guerrero, Luan T. Tran, Christoph W. Müller, Tomi Pastinen, Gustavo H. B. Maegawa, Isabelle Thiffault, Geneviève Bernard
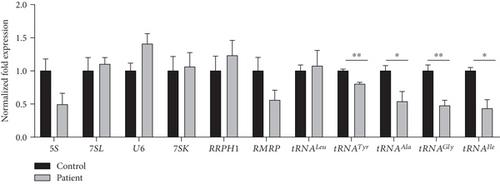
{"title":"POLR3K 中的新型致病变异导致 POLR3 相关性白营养不良症","authors":"Stefanie Perrier, Julia Macintosh, Agata D. Misiaszek, Gabrielle Lambert, Kether Guerrero, Luan T. Tran, Christoph W. Müller, Tomi Pastinen, Gustavo H. B. Maegawa, Isabelle Thiffault, Geneviève Bernard","doi":"10.1155/2024/8807171","DOIUrl":null,"url":null,"abstract":"<p>POLR3-related hypomyelinating leukodystrophy (POLR3-HLD) is a rare inherited neurological disorder caused by biallelic pathogenic variants in specific genes encoding subunits of RNA polymerase III (Pol III). Here, we report the third patient worldwide with pathogenic variants in <i>POLR3K</i> and clinical features consistent with POLR3-HLD. The female patient presented with mild intellectual and behavioural disturbances in childhood, as well as growth delay, with brain MRI revealing diffuse hypomyelination and a pattern consistent with POLR3-HLD. In adolescence, she manifested minor motor dysfunction. Next-generation sequencing revealed a paternally inherited missense variant in <i>POLR3K</i> (c.322G>T; p.D108Y) and a maternally inherited large deletion, spanning approximately 17.8 kb from chr16:30,362-48,162. The missense variant is located at the C-terminus position of the protein and is predicted to impair residue interactions and cause steric interference in enzyme conformational changes. The large deletion encompasses the third and last exon of <i>POLR3K</i>, leading to a likely amorphic truncated protein product lacking the final 42 amino acids from the total 108 amino acid–length protein. Studies of RNA-level expression showed a significant reduction in the levels of <i>POLR3K</i> RNA in the patient compared to the control. In considering whether the transcriptional function of Pol III was affected, the expression of several Pol III-transcribed RNAs was measured, where the levels of several distinct tRNAs were significantly reduced in the patient while the expression of other RNA transcripts was not decreased, suggesting that Pol III retains partial function. This study provides further evidence for the association of pathogenic variants in <i>POLR3K</i> with POLR3-HLD, expanding the spectrum of pathogenic variants in genes encoding for Pol III subunits associated with this disease.</p>","PeriodicalId":13061,"journal":{"name":"Human Mutation","volume":"2024 1","pages":""},"PeriodicalIF":3.7000,"publicationDate":"2024-07-31","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1155/2024/8807171","citationCount":"0","resultStr":"{\"title\":\"Novel Pathogenic Variants in POLR3K Cause POLR3-Related Leukodystrophy\",\"authors\":\"Stefanie Perrier, Julia Macintosh, Agata D. Misiaszek, Gabrielle Lambert, Kether Guerrero, Luan T. Tran, Christoph W. Müller, Tomi Pastinen, Gustavo H. B. Maegawa, Isabelle Thiffault, Geneviève Bernard\",\"doi\":\"10.1155/2024/8807171\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>POLR3-related hypomyelinating leukodystrophy (POLR3-HLD) is a rare inherited neurological disorder caused by biallelic pathogenic variants in specific genes encoding subunits of RNA polymerase III (Pol III). Here, we report the third patient worldwide with pathogenic variants in <i>POLR3K</i> and clinical features consistent with POLR3-HLD. The female patient presented with mild intellectual and behavioural disturbances in childhood, as well as growth delay, with brain MRI revealing diffuse hypomyelination and a pattern consistent with POLR3-HLD. In adolescence, she manifested minor motor dysfunction. Next-generation sequencing revealed a paternally inherited missense variant in <i>POLR3K</i> (c.322G>T; p.D108Y) and a maternally inherited large deletion, spanning approximately 17.8 kb from chr16:30,362-48,162. The missense variant is located at the C-terminus position of the protein and is predicted to impair residue interactions and cause steric interference in enzyme conformational changes. The large deletion encompasses the third and last exon of <i>POLR3K</i>, leading to a likely amorphic truncated protein product lacking the final 42 amino acids from the total 108 amino acid–length protein. Studies of RNA-level expression showed a significant reduction in the levels of <i>POLR3K</i> RNA in the patient compared to the control. In considering whether the transcriptional function of Pol III was affected, the expression of several Pol III-transcribed RNAs was measured, where the levels of several distinct tRNAs were significantly reduced in the patient while the expression of other RNA transcripts was not decreased, suggesting that Pol III retains partial function. This study provides further evidence for the association of pathogenic variants in <i>POLR3K</i> with POLR3-HLD, expanding the spectrum of pathogenic variants in genes encoding for Pol III subunits associated with this disease.</p>\",\"PeriodicalId\":13061,\"journal\":{\"name\":\"Human Mutation\",\"volume\":\"2024 1\",\"pages\":\"\"},\"PeriodicalIF\":3.7000,\"publicationDate\":\"2024-07-31\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1155/2024/8807171\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Human Mutation\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1155/2024/8807171\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Human Mutation","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1155/2024/8807171","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Novel Pathogenic Variants in POLR3K Cause POLR3-Related Leukodystrophy
POLR3-related hypomyelinating leukodystrophy (POLR3-HLD) is a rare inherited neurological disorder caused by biallelic pathogenic variants in specific genes encoding subunits of RNA polymerase III (Pol III). Here, we report the third patient worldwide with pathogenic variants in POLR3K and clinical features consistent with POLR3-HLD. The female patient presented with mild intellectual and behavioural disturbances in childhood, as well as growth delay, with brain MRI revealing diffuse hypomyelination and a pattern consistent with POLR3-HLD. In adolescence, she manifested minor motor dysfunction. Next-generation sequencing revealed a paternally inherited missense variant in POLR3K (c.322G>T; p.D108Y) and a maternally inherited large deletion, spanning approximately 17.8 kb from chr16:30,362-48,162. The missense variant is located at the C-terminus position of the protein and is predicted to impair residue interactions and cause steric interference in enzyme conformational changes. The large deletion encompasses the third and last exon of POLR3K, leading to a likely amorphic truncated protein product lacking the final 42 amino acids from the total 108 amino acid–length protein. Studies of RNA-level expression showed a significant reduction in the levels of POLR3K RNA in the patient compared to the control. In considering whether the transcriptional function of Pol III was affected, the expression of several Pol III-transcribed RNAs was measured, where the levels of several distinct tRNAs were significantly reduced in the patient while the expression of other RNA transcripts was not decreased, suggesting that Pol III retains partial function. This study provides further evidence for the association of pathogenic variants in POLR3K with POLR3-HLD, expanding the spectrum of pathogenic variants in genes encoding for Pol III subunits associated with this disease.
期刊介绍:
Human Mutation is a peer-reviewed journal that offers publication of original Research Articles, Methods, Mutation Updates, Reviews, Database Articles, Rapid Communications, and Letters on broad aspects of mutation research in humans. Reports of novel DNA variations and their phenotypic consequences, reports of SNPs demonstrated as valuable for genomic analysis, descriptions of new molecular detection methods, and novel approaches to clinical diagnosis are welcomed. Novel reports of gene organization at the genomic level, reported in the context of mutation investigation, may be considered. The journal provides a unique forum for the exchange of ideas, methods, and applications of interest to molecular, human, and medical geneticists in academic, industrial, and clinical research settings worldwide.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


