{"title":"基因组测序揭示了拷贝数变异在听力损失中的作用,并在 DFNB1 基因座中发现了具有创始人效应的新缺失基因","authors":"Zibin Lin, Jiale Xiang, Xiangzhong Sun, Nana Song, Xiaozhou Liu, Qinming Cai, Jing Yang, Haodong Ye, Jiangfan Xu, Hongfu Zhang, Jiguang Peng, Yu Sun, Zhiyu Peng","doi":"10.1155/2024/9517114","DOIUrl":null,"url":null,"abstract":"<p>Sensorineural hearing loss is a prevalent disorder with significant genetic involvement, which is often challenging to diagnose due to genetic heterogeneity. Exome sequencing (ES) has been a standard diagnostic tool for sensorineural hearing loss, but its limitations in detecting copy number variants (CNVs) and intronic variants have prompted the exploration of genome sequencing (GS) for improved diagnostic yield. We conducted GS on 46 hearing loss families with previously negative ES results and an additional cohort of 36 patients with a monoallelic pathogenic variant in <i>GJB2</i> (the most common deafness gene). Additionally, the impact of a previously unrecognized novel 125-kb deletion in the DFNB1 locus on <i>GJB2</i> expression was assessed using quantitative polymerase chain reaction (qPCR), and haplotype analysis was performed to characterize the deletion. GS diagnosed eight cases (17%, 8/46) in the ES-negative cohort, primarily attributed to CNVs (6/8). Notably, a previously unrecognized 125 kb deletion in the DFNB1 region was identified, affecting <i>GJB2</i> expression and characterizing it as a founder effect in East Asian. In 47 patients with a monoallelic <i>GJB2</i> variant, 15% (95% CI, 7.4%–28%) were diagnosed with DFNB1 deletions. Analysis of the gnomAD database revealed the prevalence and ethnic diversity of DFNB1 deletions, with the novel 125 kb deletion emerging as a prominent pathogenic variant in East Asian, non-Finnish European, and admixed American populations. Our study highlights the utility of GS in diagnosing sensorineural hearing loss. The identification of DFNB1 deletions underscores their significant contribution to hearing loss etiology, advocating for their inclusion in routine diagnostic testing. We propose GS as a primary genetic testing approach for patients with hearing loss, offering comprehensive genomic analysis and the potential for improved diagnostic accuracy.</p>","PeriodicalId":13061,"journal":{"name":"Human Mutation","volume":"2024 1","pages":""},"PeriodicalIF":3.7000,"publicationDate":"2024-08-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1155/2024/9517114","citationCount":"0","resultStr":"{\"title\":\"Genome Sequencing Unveils the Role of Copy Number Variants in Hearing Loss and Identifies Novel Deletions With Founder Effect in the DFNB1 Locus\",\"authors\":\"Zibin Lin, Jiale Xiang, Xiangzhong Sun, Nana Song, Xiaozhou Liu, Qinming Cai, Jing Yang, Haodong Ye, Jiangfan Xu, Hongfu Zhang, Jiguang Peng, Yu Sun, Zhiyu Peng\",\"doi\":\"10.1155/2024/9517114\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Sensorineural hearing loss is a prevalent disorder with significant genetic involvement, which is often challenging to diagnose due to genetic heterogeneity. Exome sequencing (ES) has been a standard diagnostic tool for sensorineural hearing loss, but its limitations in detecting copy number variants (CNVs) and intronic variants have prompted the exploration of genome sequencing (GS) for improved diagnostic yield. We conducted GS on 46 hearing loss families with previously negative ES results and an additional cohort of 36 patients with a monoallelic pathogenic variant in <i>GJB2</i> (the most common deafness gene). Additionally, the impact of a previously unrecognized novel 125-kb deletion in the DFNB1 locus on <i>GJB2</i> expression was assessed using quantitative polymerase chain reaction (qPCR), and haplotype analysis was performed to characterize the deletion. GS diagnosed eight cases (17%, 8/46) in the ES-negative cohort, primarily attributed to CNVs (6/8). Notably, a previously unrecognized 125 kb deletion in the DFNB1 region was identified, affecting <i>GJB2</i> expression and characterizing it as a founder effect in East Asian. In 47 patients with a monoallelic <i>GJB2</i> variant, 15% (95% CI, 7.4%–28%) were diagnosed with DFNB1 deletions. Analysis of the gnomAD database revealed the prevalence and ethnic diversity of DFNB1 deletions, with the novel 125 kb deletion emerging as a prominent pathogenic variant in East Asian, non-Finnish European, and admixed American populations. Our study highlights the utility of GS in diagnosing sensorineural hearing loss. The identification of DFNB1 deletions underscores their significant contribution to hearing loss etiology, advocating for their inclusion in routine diagnostic testing. We propose GS as a primary genetic testing approach for patients with hearing loss, offering comprehensive genomic analysis and the potential for improved diagnostic accuracy.</p>\",\"PeriodicalId\":13061,\"journal\":{\"name\":\"Human Mutation\",\"volume\":\"2024 1\",\"pages\":\"\"},\"PeriodicalIF\":3.7000,\"publicationDate\":\"2024-08-06\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1155/2024/9517114\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Human Mutation\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1155/2024/9517114\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Human Mutation","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1155/2024/9517114","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Genome Sequencing Unveils the Role of Copy Number Variants in Hearing Loss and Identifies Novel Deletions With Founder Effect in the DFNB1 Locus
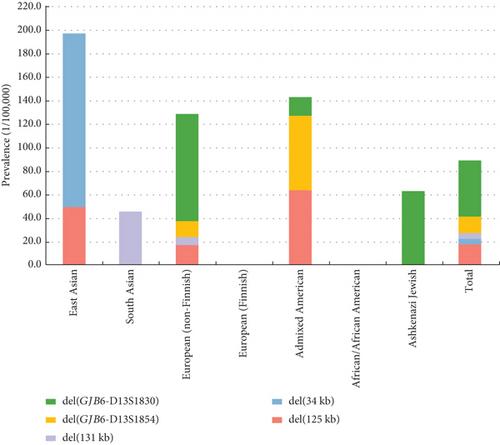
Sensorineural hearing loss is a prevalent disorder with significant genetic involvement, which is often challenging to diagnose due to genetic heterogeneity. Exome sequencing (ES) has been a standard diagnostic tool for sensorineural hearing loss, but its limitations in detecting copy number variants (CNVs) and intronic variants have prompted the exploration of genome sequencing (GS) for improved diagnostic yield. We conducted GS on 46 hearing loss families with previously negative ES results and an additional cohort of 36 patients with a monoallelic pathogenic variant in GJB2 (the most common deafness gene). Additionally, the impact of a previously unrecognized novel 125-kb deletion in the DFNB1 locus on GJB2 expression was assessed using quantitative polymerase chain reaction (qPCR), and haplotype analysis was performed to characterize the deletion. GS diagnosed eight cases (17%, 8/46) in the ES-negative cohort, primarily attributed to CNVs (6/8). Notably, a previously unrecognized 125 kb deletion in the DFNB1 region was identified, affecting GJB2 expression and characterizing it as a founder effect in East Asian. In 47 patients with a monoallelic GJB2 variant, 15% (95% CI, 7.4%–28%) were diagnosed with DFNB1 deletions. Analysis of the gnomAD database revealed the prevalence and ethnic diversity of DFNB1 deletions, with the novel 125 kb deletion emerging as a prominent pathogenic variant in East Asian, non-Finnish European, and admixed American populations. Our study highlights the utility of GS in diagnosing sensorineural hearing loss. The identification of DFNB1 deletions underscores their significant contribution to hearing loss etiology, advocating for their inclusion in routine diagnostic testing. We propose GS as a primary genetic testing approach for patients with hearing loss, offering comprehensive genomic analysis and the potential for improved diagnostic accuracy.
期刊介绍:
Human Mutation is a peer-reviewed journal that offers publication of original Research Articles, Methods, Mutation Updates, Reviews, Database Articles, Rapid Communications, and Letters on broad aspects of mutation research in humans. Reports of novel DNA variations and their phenotypic consequences, reports of SNPs demonstrated as valuable for genomic analysis, descriptions of new molecular detection methods, and novel approaches to clinical diagnosis are welcomed. Novel reports of gene organization at the genomic level, reported in the context of mutation investigation, may be considered. The journal provides a unique forum for the exchange of ideas, methods, and applications of interest to molecular, human, and medical geneticists in academic, industrial, and clinical research settings worldwide.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


