{"title":"全原子/联合原子/粗粒度混合模型与模拟退火相结合的稳健诱导拟合对接方法","authors":"Dexin Lu, Ding Luo, Yuwei Zhang* and Binju Wang*, ","doi":"10.1021/acs.jctc.4c0065310.1021/acs.jctc.4c00653","DOIUrl":null,"url":null,"abstract":"<p >Molecular docking remains an indispensable tool in computational biology and structure-based drug discovery. However, the correct prediction of binding poses remains a major challenge for molecular docking, especially for target proteins where a substrate binding induces significant reorganization of the active site. Here, we introduce an Induced Fit Docking (IFD) approach named AA/UA/CG-SA-IFD, which combines a hybrid All-Atom/United-Atom/Coarse-Grained model with Simulated Annealing. In this approach, the core region is represented by the All-Atom(AA) model, while the protein environment beyond the core region and the solvent are treated with either the United-Atom (UA) or the Coarse-Grained (CG) model. By combining the Elastic Network Model (ENM) for the CG region, the hybrid model ensures a reasonable description of ligand binding and the environmental effects of the protein, facilitating highly efficient and reliable sampling of ligand binding through Simulated Annealing (SA) at a high temperature. Upon validation with two testing sets, the AA/UA/CG-SA-IFD approach demonstrates remarkable accuracy and efficiency in induced fit docking, even for challenging cases where the docked poses significantly deviate from crystal structures.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":"20 14","pages":"6414–6423 6414–6423"},"PeriodicalIF":5.5000,"publicationDate":"2024-07-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"A Robust Induced Fit Docking Approach with the Combination of the Hybrid All-Atom/United-Atom/Coarse-Grained Model and Simulated Annealing\",\"authors\":\"Dexin Lu, Ding Luo, Yuwei Zhang* and Binju Wang*, \",\"doi\":\"10.1021/acs.jctc.4c0065310.1021/acs.jctc.4c00653\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Molecular docking remains an indispensable tool in computational biology and structure-based drug discovery. However, the correct prediction of binding poses remains a major challenge for molecular docking, especially for target proteins where a substrate binding induces significant reorganization of the active site. Here, we introduce an Induced Fit Docking (IFD) approach named AA/UA/CG-SA-IFD, which combines a hybrid All-Atom/United-Atom/Coarse-Grained model with Simulated Annealing. In this approach, the core region is represented by the All-Atom(AA) model, while the protein environment beyond the core region and the solvent are treated with either the United-Atom (UA) or the Coarse-Grained (CG) model. By combining the Elastic Network Model (ENM) for the CG region, the hybrid model ensures a reasonable description of ligand binding and the environmental effects of the protein, facilitating highly efficient and reliable sampling of ligand binding through Simulated Annealing (SA) at a high temperature. Upon validation with two testing sets, the AA/UA/CG-SA-IFD approach demonstrates remarkable accuracy and efficiency in induced fit docking, even for challenging cases where the docked poses significantly deviate from crystal structures.</p>\",\"PeriodicalId\":45,\"journal\":{\"name\":\"Journal of Chemical Theory and Computation\",\"volume\":\"20 14\",\"pages\":\"6414–6423 6414–6423\"},\"PeriodicalIF\":5.5000,\"publicationDate\":\"2024-07-05\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Theory and Computation\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jctc.4c00653\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jctc.4c00653","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
A Robust Induced Fit Docking Approach with the Combination of the Hybrid All-Atom/United-Atom/Coarse-Grained Model and Simulated Annealing
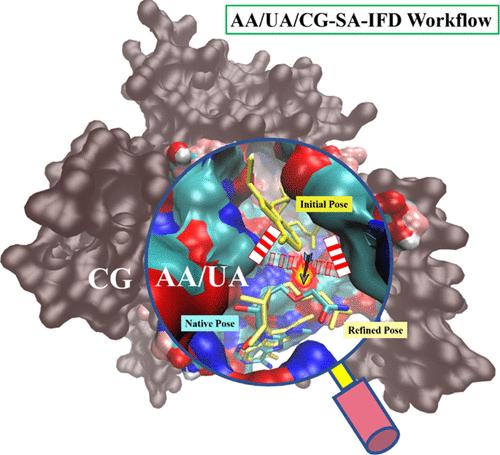
Molecular docking remains an indispensable tool in computational biology and structure-based drug discovery. However, the correct prediction of binding poses remains a major challenge for molecular docking, especially for target proteins where a substrate binding induces significant reorganization of the active site. Here, we introduce an Induced Fit Docking (IFD) approach named AA/UA/CG-SA-IFD, which combines a hybrid All-Atom/United-Atom/Coarse-Grained model with Simulated Annealing. In this approach, the core region is represented by the All-Atom(AA) model, while the protein environment beyond the core region and the solvent are treated with either the United-Atom (UA) or the Coarse-Grained (CG) model. By combining the Elastic Network Model (ENM) for the CG region, the hybrid model ensures a reasonable description of ligand binding and the environmental effects of the protein, facilitating highly efficient and reliable sampling of ligand binding through Simulated Annealing (SA) at a high temperature. Upon validation with two testing sets, the AA/UA/CG-SA-IFD approach demonstrates remarkable accuracy and efficiency in induced fit docking, even for challenging cases where the docked poses significantly deviate from crystal structures.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


