对 "从 Ab Initio 预生物化学逐步合成 Strecker 氨基酸 "的更正
IF 4.6
2区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
修正自由能曲线后的新反应图。颜色代码与图 E.4 一致:绿色部分是为了检查原论文中遗漏的 vdW 修正的影响而重新制作的;(1)红色部分是关于滞后修正的部分,我们必须为其重新定义新的 s12 和 z12 坐标。本文引用了其他 5 篇文章。本文尚未被其他出版物引用。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Correction to “Step by Step Strecker Amino Acid Synthesis from Ab Initio Prebiotic Chemistry”
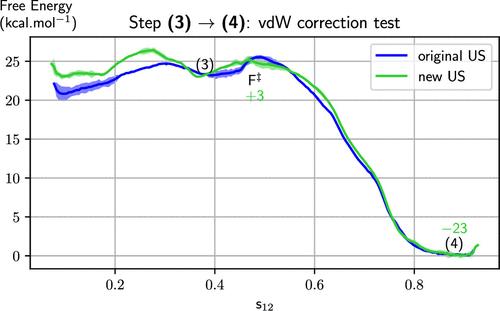
In the cited work, (1) Grimme’s van der Waals (vdW) corrections (2) were accidentally overlooked and not included in the potential calculations. Our goal is to address this oversight to prevent its recurrence in future studies. The oversight was originated from the vdW section of the CPMD software input file, with no warning in version 4.1 of the CPMD software. (3) The keywords ”EMPIRICAL CORRECTION” and ”END EMPIRICAL CORRECTION” are missing in the description of the vdW section of the CPMD manual. To evaluate how this oversight initially impacted the article’s conclusions, we performed a new umbrella sampling (US) simulation for the transition from step (3) to (4) using the corrected input file. We chose this step for two reasons: it depends on a long-range interaction between two reactive molecules that could be influenced by the omission of vdW corrections, and it has been used as a reliable benchmark reaction for additional simulations. (4) The protocol for the revised (3) → (4) US simulation remained the same as in the ref (1) in each window we generated 14 ps of trajectory, divided into 7 ps for equilibration and 7 ps for free-energy sampling. The results, shown in Figure E.1, indicate consistency between the two calculations, with ΔrF = −23 ± 0.5 kcal.mol–1 for both. As anticipated for condensed phase chemical reactions, empirical vdW corrections have minimal impact on the results. Additionally, we confirmed that the corrected input file accurately implements Grimme’s van der Waals D2 corrections and that these are properly included in the force estimates. We expect that incorporating vdW corrections in other steps of the mechanism will still yield results within the error margins of the initial study. (1) Figure E.1. Results of the new US simulation with Grimme’s D2 correction. The new free-energy curve is depicted in green, while the original one, as presented in the article, is presented in blue. The reactant position for the step s(3)→(4) is located at 0.4 on the s12 axis. The error bars are indicated for both with shaded areas. van der Waals corrections are consistently accounted for also in the simulations described in the following sections. An issue was identified during the sampling of the (2) → (3) step in the Strecker mechanism, as described in ref (1), which involves the formation of a cation through the elimination of a water molecule. A pronounced gradient was noted in a specific region of the free-energy curve. After incorporating additional US windows in this region, the free-energy landscape was impacted, increasing the final free-energy difference between reactants and products by 4 kcal·mol–1, as shown in Figure E.2. The s12 coordinate initially designed for this step seems inadequate to capture this chemical transformation, thus requiring a further determination of the free-energy curve. Consequently, we defined a new set of coordinates, s12 and z12, based on new committor analysis and proceeded to repeat the entire US simulation for this step to verify the accuracy of the results. Figure E.2. Effect of the addition of a US window on the (2) → (3) step. The blue curve, as presented in the article, represents the original data, while the red curve includes the effect of an additional window. After carrying out a detailed analysis of the US data, we spotted hysteresis effects that were initially overlooked, as shown in Figure E.3. These effects were evidenced through the determination of new collective variables and a continuity check, as described in our protocol paper. (5) For each step, we established new pairs of s12 and z12 variables and repeated the US simulations. Notably, Step (1) → (2’) was subdivided into two distinct processes: the HCN deprotonation (1) → (1’) and the amine addition (1’) → (2’). The deprotonation reaction coordinate was defined by the coordination of the carbon with hydrogen, which underlines the relative simplicity of the reaction. Figure E.3. Hysteresis has been observed in previous US simulations at several steps: a) In step (1) → (2’), hysteresis was detected in the protonation of the cyanide’s carbon, cC2(H). b) In step (2) → (3), the critical variable is the protonation of the leaving oxygen, cO66(H). c) For step (4) → (5′), the critical variable involves the protonation of the leaving nitrogen, cN85(H). d) In step (5′) → (5), the critical variable is again the protonation of the leaving nitrogen, cN85(H). The s12 coordinate is used as the horizontal axis for each graph. The new, revised free-energy curves for this process are reported in Figure E.4. The updated diagram is presented in Figure E.5. We have not identified additional errors of this nature in the other steps. Figure E.4. Free-energy curves calculated out of the newly performed US simulations, with error bars. The one made to check vdW correction effects is represented in green. The ones performed to correct the observed hysteresis in the original data are drawn in red. Figure E.5. New reaction diagram after the correction of the free-energy curves. The color code is consistent with Figure E.4: in green the section remade to check effect of vdW corrections omission in the original paper, (1) in red the sections that regard hysteresis corrections for which we had to redefine new s12 and z12 coordinates. This article references 5 other publications. This article has not yet been cited by other publications.
The keywords ”EMPIRICAL CORRECTION” and ”END EMPIRICAL CORRECTION” are missing in the description of the vdW section of the CPMD manual. To evaluate how this oversight initially impacted the article’s conclusions, we performed a new umbrella sampling (US) simulation for the transition from step (3) to (4) using the corrected input file. We chose this step for two reasons: it depends on a long-range interaction between two reactive molecules that could be influenced by the omission of vdW corrections, and it has been used as a reliable benchmark reaction for additional simulations. (4) The protocol for the revised (3) → (4) US simulation remained the same as in the ref (1) in each window we generated 14 ps of trajectory, divided into 7 ps for equilibration and 7 ps for free-energy sampling. The results, shown in Figure E.1, indicate consistency between the two calculations, with ΔrF = −23 ± 0.5 kcal.mol–1 for both. As anticipated for condensed phase chemical reactions, empirical vdW corrections have minimal impact on the results. Additionally, we confirmed that the corrected input file accurately implements Grimme’s van der Waals D2 corrections and that these are properly included in the force estimates. We expect that incorporating vdW corrections in other steps of the mechanism will still yield results within the error margins of the initial study. (1) Figure E.1. Results of the new US simulation with Grimme’s D2 correction. The new free-energy curve is depicted in green, while the original one, as presented in the article, is presented in blue. The reactant position for the step s(3)→(4) is located at 0.4 on the s12 axis. The error bars are indicated for both with shaded areas. van der Waals corrections are consistently accounted for also in the simulations described in the following sections. An issue was identified during the sampling of the (2) → (3) step in the Strecker mechanism, as described in ref (1), which involves the formation of a cation through the elimination of a water molecule. A pronounced gradient was noted in a specific region of the free-energy curve. After incorporating additional US windows in this region, the free-energy landscape was impacted, increasing the final free-energy difference between reactants and products by 4 kcal·mol–1, as shown in Figure E.2. The s12 coordinate initially designed for this step seems inadequate to capture this chemical transformation, thus requiring a further determination of the free-energy curve. Consequently, we defined a new set of coordinates, s12 and z12, based on new committor analysis and proceeded to repeat the entire US simulation for this step to verify the accuracy of the results. Figure E.2. Effect of the addition of a US window on the (2) → (3) step. The blue curve, as presented in the article, represents the original data, while the red curve includes the effect of an additional window. After carrying out a detailed analysis of the US data, we spotted hysteresis effects that were initially overlooked, as shown in Figure E.3. These effects were evidenced through the determination of new collective variables and a continuity check, as described in our protocol paper. (5) For each step, we established new pairs of s12 and z12 variables and repeated the US simulations. Notably, Step (1) → (2’) was subdivided into two distinct processes: the HCN deprotonation (1) → (1’) and the amine addition (1’) → (2’). The deprotonation reaction coordinate was defined by the coordination of the carbon with hydrogen, which underlines the relative simplicity of the reaction. Figure E.3. Hysteresis has been observed in previous US simulations at several steps: a) In step (1) → (2’), hysteresis was detected in the protonation of the cyanide’s carbon, cC2(H). b) In step (2) → (3), the critical variable is the protonation of the leaving oxygen, cO66(H). c) For step (4) → (5′), the critical variable involves the protonation of the leaving nitrogen, cN85(H). d) In step (5′) → (5), the critical variable is again the protonation of the leaving nitrogen, cN85(H). The s12 coordinate is used as the horizontal axis for each graph. The new, revised free-energy curves for this process are reported in Figure E.4. The updated diagram is presented in Figure E.5. We have not identified additional errors of this nature in the other steps. Figure E.4. Free-energy curves calculated out of the newly performed US simulations, with error bars. The one made to check vdW correction effects is represented in green. The ones performed to correct the observed hysteresis in the original data are drawn in red. Figure E.5. New reaction diagram after the correction of the free-energy curves. The color code is consistent with Figure E.4: in green the section remade to check effect of vdW corrections omission in the original paper, (1) in red the sections that regard hysteresis corrections for which we had to redefine new s12 and z12 coordinates. This article references 5 other publications. This article has not yet been cited by other publications.
The keywords ”EMPIRICAL CORRECTION” and ”END EMPIRICAL CORRECTION” are missing in the description of the vdW section of the CPMD manual. To evaluate how this oversight initially impacted the article’s conclusions, we performed a new umbrella sampling (US) simulation for the transition from step (3) to (4) using the corrected input file. We chose this step for two reasons: it depends on a long-range interaction between two reactive molecules that could be influenced by the omission of vdW corrections, and it has been used as a reliable benchmark reaction for additional simulations. (4) The protocol for the revised (3) → (4) US simulation remained the same as in the ref (1) in each window we generated 14 ps of trajectory, divided into 7 ps for equilibration and 7 ps for free-energy sampling. The results, shown in Figure E.1, indicate consistency between the two calculations, with ΔrF = −23 ± 0.5 kcal.mol–1 for both. As anticipated for condensed phase chemical reactions, empirical vdW corrections have minimal impact on the results. Additionally, we confirmed that the corrected input file accurately implements Grimme’s van der Waals D2 corrections and that these are properly included in the force estimates. We expect that incorporating vdW corrections in other steps of the mechanism will still yield results within the error margins of the initial study. (1) Figure E.1. Results of the new US simulation with Grimme’s D2 correction. The new free-energy curve is depicted in green, while the original one, as presented in the article, is presented in blue. The reactant position for the step s(3)→(4) is located at 0.4 on the s12 axis. The error bars are indicated for both with shaded areas. van der Waals corrections are consistently accounted for also in the simulations described in the following sections. An issue was identified during the sampling of the (2) → (3) step in the Strecker mechanism, as described in ref (1), which involves the formation of a cation through the elimination of a water molecule. A pronounced gradient was noted in a specific region of the free-energy curve. After incorporating additional US windows in this region, the free-energy landscape was impacted, increasing the final free-energy difference between reactants and products by 4 kcal·mol–1, as shown in Figure E.2. The s12 coordinate initially designed for this step seems inadequate to capture this chemical transformation, thus requiring a further determination of the free-energy curve. Consequently, we defined a new set of coordinates, s12 and z12, based on new committor analysis and proceeded to repeat the entire US simulation for this step to verify the accuracy of the results. Figure E.2. Effect of the addition of a US window on the (2) → (3) step. The blue curve, as presented in the article, represents the original data, while the red curve includes the effect of an additional window. After carrying out a detailed analysis of the US data, we spotted hysteresis effects that were initially overlooked, as shown in Figure E.3. These effects were evidenced through the determination of new collective variables and a continuity check, as described in our protocol paper. (5) For each step, we established new pairs of s12 and z12 variables and repeated the US simulations. Notably, Step (1) → (2’) was subdivided into two distinct processes: the HCN deprotonation (1) → (1’) and the amine addition (1’) → (2’). The deprotonation reaction coordinate was defined by the coordination of the carbon with hydrogen, which underlines the relative simplicity of the reaction. Figure E.3. Hysteresis has been observed in previous US simulations at several steps: a) In step (1) → (2’), hysteresis was detected in the protonation of the cyanide’s carbon, cC2(H). b) In step (2) → (3), the critical variable is the protonation of the leaving oxygen, cO66(H). c) For step (4) → (5′), the critical variable involves the protonation of the leaving nitrogen, cN85(H). d) In step (5′) → (5), the critical variable is again the protonation of the leaving nitrogen, cN85(H). The s12 coordinate is used as the horizontal axis for each graph. The new, revised free-energy curves for this process are reported in Figure E.4. The updated diagram is presented in Figure E.5. We have not identified additional errors of this nature in the other steps. Figure E.4. Free-energy curves calculated out of the newly performed US simulations, with error bars. The one made to check vdW correction effects is represented in green. The ones performed to correct the observed hysteresis in the original data are drawn in red. Figure E.5. New reaction diagram after the correction of the free-energy curves. The color code is consistent with Figure E.4: in green the section remade to check effect of vdW corrections omission in the original paper, (1) in red the sections that regard hysteresis corrections for which we had to redefine new s12 and z12 coordinates. This article references 5 other publications. This article has not yet been cited by other publications.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

The Journal of Physical Chemistry Letters
CHEMISTRY, PHYSICAL-NANOSCIENCE & NANOTECHNOLOGY
CiteScore
9.60
自引率
7.00%
发文量
1519
审稿时长
1.6 months
期刊介绍:
The Journal of Physical Chemistry (JPC) Letters is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, chemical physicists, physicists, material scientists, and engineers. An important criterion for acceptance is that the paper reports a significant scientific advance and/or physical insight such that rapid publication is essential. Two issues of JPC Letters are published each month.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


