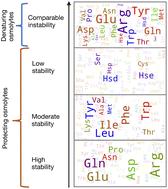
{"title":"渗透溶质诱导蛋白质稳定性的溶剂可及表面积评估分子基础。","authors":"Shampa Raghunathan","doi":"10.1039/D4RA02576H","DOIUrl":null,"url":null,"abstract":"<p >In solvent-modulated protein folding, under certain physiological conditions, an equilibrium exists between the unfolded and folded states of the protein without any need to break or make a covalent bond. In this process, interactions between various protein groups (peptides) and solvent molecules are known to play a major role in determining the directionality of the chemical reaction. However, an understanding of the mechanism of action of the co(solvent) by a generic theoretical underpinning is lacking. In this study, a generic solvation model is developed based on statistical mechanics and the thermodynamic transfer free energy model by considering the microenvironment polarity of the interacting co(solvent)–protein system. According to this model, polarity and the fractional solvent-accessible surface areas contribute to the interaction energies. The present model includes various orientations of participating interactant solvent surfaces of suitable areas. As model systems, besides the backbone we consider naturally occurring amino acid residues solvated in ten different osmolytes, small organic compounds known to modulate protein stability. The present model is able to predict the correct trend of the osmolyte–peptide interactions ranging from stabilizing to destabilizing not only for the backbone but also for side chains. Our model predicts Asn, Gln, Asp, Glu, Arg and Pro to be highly stable in most of the protecting osmolytes while Ala, Val, Ile, Leu, Thr, Met, Lys, Phe, Trp and Tyr are predicted to be moderately stable, and Ser, Cys and Histidine are predicted to be least stable. However, in denaturing solvents, both backbone and side chain models show similar stabilities in urea and guanidine. One of the important aspects of this model is that it is essentially parameter-free and consistent with the electrostatics of the interaction partners that make this model suitable for estimating any solute–solvent interaction energies.</p>","PeriodicalId":102,"journal":{"name":"RSC Advances","volume":" 34","pages":" 25031-25041"},"PeriodicalIF":4.6000,"publicationDate":"2024-08-09","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11310836/pdf/","citationCount":"0","resultStr":"{\"title\":\"Solvent accessible surface area-assessed molecular basis of osmolyte-induced protein stability†\",\"authors\":\"Shampa Raghunathan\",\"doi\":\"10.1039/D4RA02576H\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >In solvent-modulated protein folding, under certain physiological conditions, an equilibrium exists between the unfolded and folded states of the protein without any need to break or make a covalent bond. In this process, interactions between various protein groups (peptides) and solvent molecules are known to play a major role in determining the directionality of the chemical reaction. However, an understanding of the mechanism of action of the co(solvent) by a generic theoretical underpinning is lacking. In this study, a generic solvation model is developed based on statistical mechanics and the thermodynamic transfer free energy model by considering the microenvironment polarity of the interacting co(solvent)–protein system. According to this model, polarity and the fractional solvent-accessible surface areas contribute to the interaction energies. The present model includes various orientations of participating interactant solvent surfaces of suitable areas. As model systems, besides the backbone we consider naturally occurring amino acid residues solvated in ten different osmolytes, small organic compounds known to modulate protein stability. The present model is able to predict the correct trend of the osmolyte–peptide interactions ranging from stabilizing to destabilizing not only for the backbone but also for side chains. Our model predicts Asn, Gln, Asp, Glu, Arg and Pro to be highly stable in most of the protecting osmolytes while Ala, Val, Ile, Leu, Thr, Met, Lys, Phe, Trp and Tyr are predicted to be moderately stable, and Ser, Cys and Histidine are predicted to be least stable. However, in denaturing solvents, both backbone and side chain models show similar stabilities in urea and guanidine. One of the important aspects of this model is that it is essentially parameter-free and consistent with the electrostatics of the interaction partners that make this model suitable for estimating any solute–solvent interaction energies.</p>\",\"PeriodicalId\":102,\"journal\":{\"name\":\"RSC Advances\",\"volume\":\" 34\",\"pages\":\" 25031-25041\"},\"PeriodicalIF\":4.6000,\"publicationDate\":\"2024-08-09\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11310836/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"RSC Advances\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2024/ra/d4ra02576h\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"RSC Advances","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2024/ra/d4ra02576h","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
摘要
在溶剂调节的蛋白质折叠过程中,在某些生理条件下,蛋白质的展开态和折叠态之间存在着一种平衡状态,无需破坏或制造共价键。众所周知,在这一过程中,各种蛋白质基团(肽)和溶剂分子之间的相互作用在决定化学反应的方向性方面发挥着重要作用。然而,目前还缺乏通过通用理论基础来理解共(溶剂)作用机制的方法。本研究以统计力学和热力学传递自由能模型为基础,通过考虑相互作用的助(溶剂)-蛋白质系统的微环境极性,建立了一个通用溶解模型。根据该模型,极性和部分溶剂可及表面积对相互作用能有贡献。本模型包括参与相互作用的溶剂表面在适当面积上的各种取向。作为模型系统,除了骨架外,我们还考虑了溶解在十种不同渗透溶质(已知可调节蛋白质稳定性的小型有机化合物)中的天然氨基酸残基。本模型能够预测渗透溶质与肽相互作用的正确趋势,从稳定到不稳定,不仅针对骨架,也针对侧链。根据我们的模型预测,Asn、Gln、Asp、Glu、Arg 和 Pro 在大多数保护性渗透溶液中高度稳定,而 Ala、Val、Ile、Leu、Thr、Met、Lys、Phe、Trp 和 Tyr 中度稳定,Ser、Cys 和组氨酸最不稳定。不过,在变性溶剂中,骨架和侧链模型在尿素和胍中的稳定性相似。该模型的一个重要方面是它基本上不需要参数,并且与相互作用伙伴的静电一致,这使得该模型适用于估算任何溶质-溶剂相互作用的能量。
Solvent accessible surface area-assessed molecular basis of osmolyte-induced protein stability†
In solvent-modulated protein folding, under certain physiological conditions, an equilibrium exists between the unfolded and folded states of the protein without any need to break or make a covalent bond. In this process, interactions between various protein groups (peptides) and solvent molecules are known to play a major role in determining the directionality of the chemical reaction. However, an understanding of the mechanism of action of the co(solvent) by a generic theoretical underpinning is lacking. In this study, a generic solvation model is developed based on statistical mechanics and the thermodynamic transfer free energy model by considering the microenvironment polarity of the interacting co(solvent)–protein system. According to this model, polarity and the fractional solvent-accessible surface areas contribute to the interaction energies. The present model includes various orientations of participating interactant solvent surfaces of suitable areas. As model systems, besides the backbone we consider naturally occurring amino acid residues solvated in ten different osmolytes, small organic compounds known to modulate protein stability. The present model is able to predict the correct trend of the osmolyte–peptide interactions ranging from stabilizing to destabilizing not only for the backbone but also for side chains. Our model predicts Asn, Gln, Asp, Glu, Arg and Pro to be highly stable in most of the protecting osmolytes while Ala, Val, Ile, Leu, Thr, Met, Lys, Phe, Trp and Tyr are predicted to be moderately stable, and Ser, Cys and Histidine are predicted to be least stable. However, in denaturing solvents, both backbone and side chain models show similar stabilities in urea and guanidine. One of the important aspects of this model is that it is essentially parameter-free and consistent with the electrostatics of the interaction partners that make this model suitable for estimating any solute–solvent interaction energies.
期刊介绍:
An international, peer-reviewed journal covering all of the chemical sciences, including multidisciplinary and emerging areas. RSC Advances is a gold open access journal allowing researchers free access to research articles, and offering an affordable open access publishing option for authors around the world.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


